Abstract
Summary: Urea cycle defect is an inborn error of ammonium metabolism caused by a deficient activity of the enzymes involved in urea synthesis. Localized short-TE proton MR spectroscopy, performed in two infants who had citrullinemia and ornithine transcarbamylase deficiency, respectively, showed a prominent increase of glutamine/glutamate and lipid/lactate complex in both cases. N-acetylaspartate, total creatine, and myo-inositol were decreased in the infant with citrullinemia. Proton MR spectroscopy provided useful information for the diagnosis and understanding of the pathophysiology of urea cycle enzyme defect.
Adequate activities of five different types of enzymes are necessary to convert excessive ammonium into urea, which is excreted into the urine (1). In ornithine transcarbamylase (OTC) deficiency, the most common form of urea cycle enzyme defect, failure of the condensation of ornithine with carbamyl phosphate leads to high plasma ammonium and high urine orotic acid levels. In citrullinemia, a less common form caused by a deficient activity of L-argininosuccinate synthetase, failure of the condensation of citrulline with aspartate leads to high plasma ammonium and high plasma citrulline levels.
Case Reports
Case 1
A male baby who was delivered by cesarean section after a full-term pregnancy was well until the third day of life when he became lethargic and hypotonic. His respiration became shallow and progressed to apnea, requiring mechanical ventilation. On the third day of life, the plasma ammonium was 3044 μmol/L (normal, 18–54 μmol/L). While the results of plasma amino acid and urine organic acid analyses were awaited, peritoneal dialysis was started; the plasma ammonium had decreased to 533 μmol/L on the fifth day but had not normalized. Because the patient remained in deep coma with no evidence of spontaneous respiration, life support systems were discontinued after consultation with the family. The infant died on the seventh day. Plasma amino acid analysis revealed a glutamine level of 4047 μmol/L (normal, 376–709 μmol/L), an alanine level of 2626 μmol/L (normal, 131–710 μmol/L), normal citrulline, and no detectable arginine. In the urine sample, the excretion of orotic acid was massively elevated and the concentration was measured as 3328 mmol/mol creatinine (normal, 0.2–6.0). Laboratory findings were consistent with OTC deficiency.
Brain MR imaging and spectroscopy were performed on the sixth day by using a 1.5-T system. Diffuse cerebral edema was suspected on T2-weighted MR images (Fig 1A and B). After MR imaging, proton MR spectra were recorded in right parietal white matter, parietal gray matter, and right basal ganglia with use of single-voxel stimulated echo acquisition mode (STEAM) sequence (3000/30/13.7/96 [TR/TE/mixing time/excitations], spectral width = 2500 Hz, number of points = 2048, voxel size = 1.8–2 cm × 2 cm × 2 cm). The obtained raw data were post-processed by water-referenced correction of the eddy current effect (2), Lorenz to Gauss transformation, Gaussian line broadening of 0.5 Hz, zerofilling of 8K, Fourier transformation, and zero-order phasing of the transformed spectrum. Major peaks at 2.01, 3.03, 3.22, and 3.56 ppm were assigned to N-acetylaspartate (NAA), creatine and phosphocreatine (total Cr), choline-containing compounds (Cho), and myo-inositol, respectively (3). Complex peaks at 2.05–2.55 ppm and 3.68–3.85 ppm were assigned to glutamine/glutamate complex and broad peaks at 0.8–1.5 ppm were assigned to lipid/lactate complex (3). Compared with an age-matched control subject, marked increases of the glutamate/glutamine and lipid/lactate complexes were observed in the spectra obtained from the patient (Fig 1C–E).

A neonate with OTC deficiency (6 days).
A and B, Axial T2-weighted (3000/102) images obtained with a fast spin-echo sequence and a 16-cm field of view, 256 × 192 matrix, and 5-mm contiguous sections, with the voxel locations and voxel sizes (7.2–8.0 cm3) as indicated for proton MR spectroscopy (STEAM sequence 3000/30).
C, Proton MR spectrum obtained from the parietal gray matter of an age-matched control subject (full-term neonate, 5 days). Glx = glutamine/glutamate complex, tCr = total Cr, mI = myo-inositol, other abbreviations are the same as used in the text.
D, Proton MR spectrum obtained from the parietal gray matter of the patient shows a prominent increase of glutamine/glutamate and lipid/lactate complexes, and relatively well preserved NAA, tCr, Cho, and mI as compared with normal spectrum.
E, Proton MR spectrum obtained from the basal ganglia of the patient also shows similar findings; a prominent increase of glutamine/glutamate and lipid/lactate complexes
Case 2
A female baby who was born after a full-term pregnancy and spontaneous vaginal delivery was well during the first 43 days but then was noted to be febrile and irritable. She was brought to her local emergency room and referred to our hospital because of hyperammonemia (673 μmol/L), uncontrollable generalized tonic-clonic seizure, and a suspected infarction in the right cerebral hemisphere on CT scan. On admission, the plasma ammonium level was 535 μmol/L (normal, 18–54 μmol/L). While the results of plasma amino acid and urine organic acid analyses were awaited, peritoneal dialysis was started; the plasma ammonium had decreased to 50–60 μmol/L after 3 days. Her consciousness level had improved and her seizure activity had subsided slowly over several days. Plasma amino acid analysis revealed a citrulline level of 1376 μmol/L (normal, 3–35 μmol/L), normal glutamine, and no detectable arginine. In the urine sample, the excretion of orotic acid was mildly elevated on the qualitative analysis. Under the diagnosis of citrullinemia, she was treated with protein-restricted milk feeding and oral administration of sodium benzoate. Over the following months, she showed a developmental delay, spastic quadriparesis, and intermittent hyperammonemic attacks, which were treated by peritoneal dialysis.
Brain MR imaging and spectroscopy were performed on the third day after admission. Proton MR spectra were recorded in both parietal white matters and in the area near the right basal ganglia, and post-processed using the same technique as described in case 1 (Fig 2A). Compared with an age-matched control subject, the spectra obtained from the patient showed marked increases of glutamate/glutamine and lipid/lactate complexes, and decrease of NAA, total Cr, and myo-inositol (Fig 2B and C). Follow-up MR imaging performed at the age of 10 months showed severe atrophy and cystic encephalomalacia in both cerebral hemispheres (Fig 2D).

{kind=link}
{kind=link}
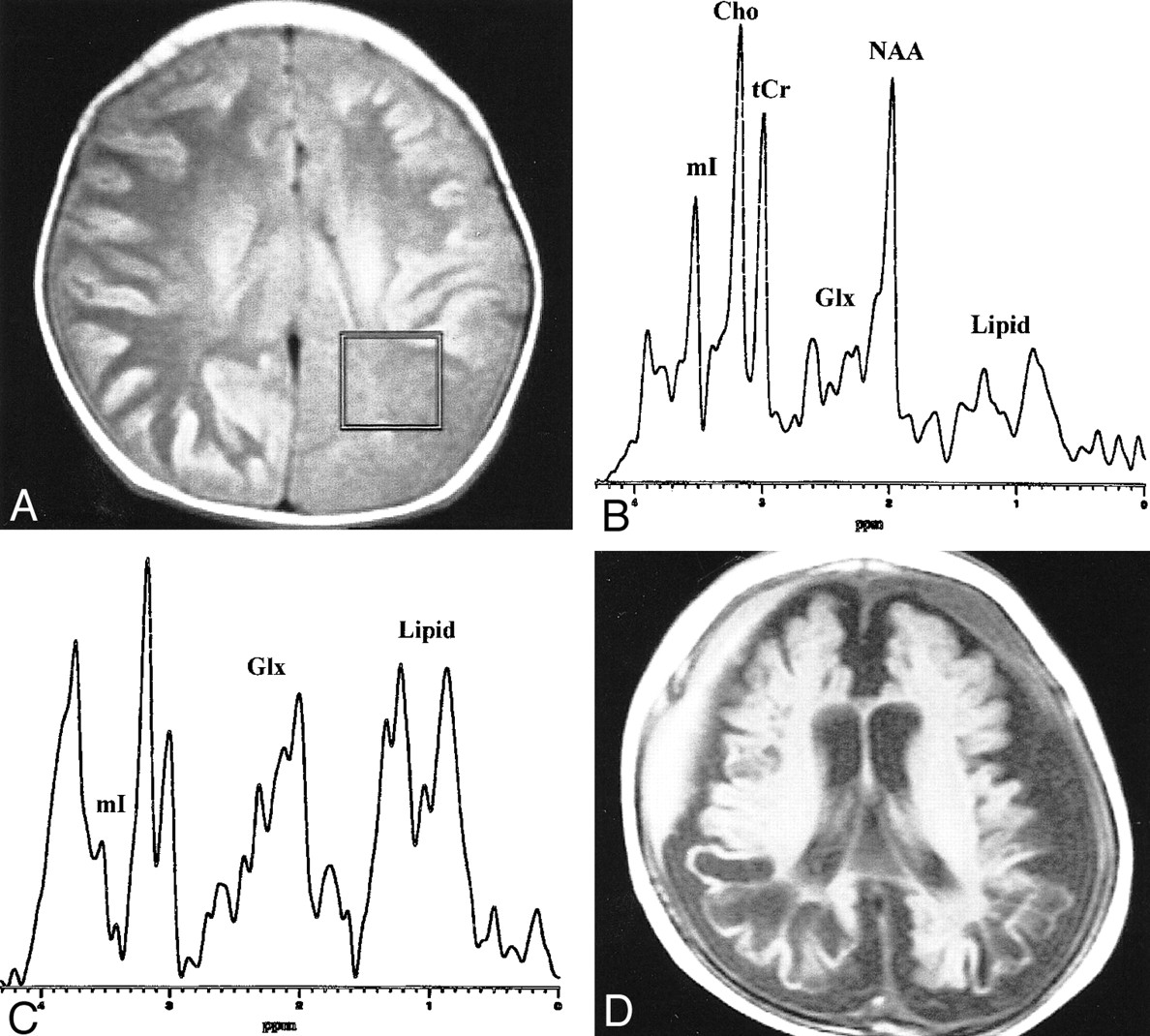
An infant with citrullinemia (57 days).
A, Axial T1-weighted (600/16) image obtained with a spin-echo sequence and a 20-cm field of view, 256 × 192 matrix, and 5-mm contiguous sections, with the voxel location and voxel size (7.2 cm3) as indicated for proton MR spectroscopy (STEAM sequence 3000/30).
B, Proton MR spectrum obtained from the parietal white matter of an age-matched control subject (2 months). Glx = glutamine/glutamate complex, tCr = total Cr, mI = myo-inositol, other abbreviations are the same as used in the text.
C, Proton MR spectrum obtained from the parietal white matter of the patient shows a prominent increase of glutamine/glutamate and lipid/lactate complexes, and a decrease of NAA, tCr, and mI as compared with normal spectrum. The amplitude of the Cho peak is relatively well preserved.
D, A follow-up axial T1-weighted (490/14) image obtained at the age of 10 months shows severe atrophy and cystic encephalomalacia in both cerebral hemispheres
Discussion
The most characteristic feature of proton MR spectroscopy in our cases was markedly increased peak intensities at 2.05–2.55 and 3.68–3.85 ppm, which were assigned to glutamate/glutamine complex. Although it is difficult to differentiate glutamine from glutamate in proton MR spectra obtained at 1.5 T, most of the increased peak intensities seem to be caused by increased cerebral glutamine concentration when considering the hyperammonemia in both patients. In hyperammonemic states, excess brain ammonia is converted to glutamine by the enzyme glutamine synthetase within astrocytes (4). The results of in vivo proton MR spectroscopy in rats during acute hyperammonemia confirmed a significant increase in cerebral glutamine concentration and a decrease in glutamate concentration (5). Connelly et al (6) also reported the increase of cerebral glutamine in two young children who had OTC deficiency by using spin-echo MR spectroscopy with long echo time (135 ms). In this study, we used a STEAM sequence with short echo time (30 ms), which has an advantage in detecting complex phase-modulated peaks of glutamine. In our cases that showed a massive increase of cerebral glutamine on proton MR spectroscopy, the clinical outcomes were poor (spastic quadriparesis or death). There is evidence that glutamine synthesis is an essential step in the development of hyperammonemic encephalopathy and glutamine is a more important factor for brain damage rather than ammonia itself (7). The increase of cerebral glutamine was reported in other hyperammonemic encephalopathies, such as chronic hepatic encephalopathies (8) or Reye's syndrome (9).
Another remarkable finding was the increase of broad peak intensities at 0.8–1.5 ppm, which were assigned to the lipid/lactate complex. Although it is difficult to differentiate lipid with lactate clearly in the short echo-time proton MR spectra, most of the broad peak intensities at 0.8–1.5 ppm may be contributed from increased mobile lipids within the investigated regions. It seems unlikely that this finding is caused by fat contamination from the scalp or bone marrow because it was observed even in the spectra obtained from the basal ganglia, where the volume of interest was far from the scalp or bone marrow fat (Fig 1B). We speculate that the increase of mobile lipids may be caused by neuroglial cell membrane damage or disintegration due to severe metabolic stress, and may be a sign foretelling significant brain damage.
A decrease of myo-inositol at 3.56 ppm was observed in the infant with citrullinemia (Fig 2C), in agreement with reported proton MR spectroscopy findings in other hyperammonemic encephalopathies, such as hepatic encephalopathy or Reye's syndrome (8, 9). In the infant who had OTC deficiency that progressed rapidly within several days, myo-inositol at 3.56 ppm was apparently well preserved, despite a massive increase of glutamine (Fig 1D and E). The apparent preservation of myo-inositol in this patient can be explained in part by several factors, such as sloping baseline, some water suppression artifacts, and the increase of glutamine/glutamate complex at 3.68–3.85 ppm. Despite these considerations, however, it is our impression that a substantial portion of the peak intensity at 3.56 ppm seems to be contributed by myo-inositol itself. This contribution seems disproportionately large compared with the massive increase of glutamine, considering the theory of myo-inositol as an intracellular osmolyte (8, 10). NAA, total Cr, and Cho were relatively well preserved in the neonate who had rapid progression of OTC deficiency. However, in the infant who had citrullinemia that progressed over several weeks, NAA and total Cr decreased. In the latter case, the decrease of NAA and total Cr may reflect neuro-axonal damage and loss of the creatine pool by cellular disintegration, respectively.
In summary, localized short–TE proton MR spectroscopy, performed in two infants who had citrullinemia and OTC deficiency, respectively, detected a prominent increase of glutamine/glutamate and lipid/lactate complexes in both cases, and a decrease of NAA, total Cr, and myo-inositol in the infant with citrullinemia. If proton MR spectroscopy shows a prominent increase of cerebral glutamine/glutamate complex in an infant, one should consider the possibility of hyperammonemic encephalopathies such as urea cycle defect or Reye's syndrome.
Footnotes
1 Presented at the annual meeting of American Society of Neuroradiology, Atlanta, GA, April 2000.
2 Address reprint requests to Choong-Gon Choi, MD, Department of Radiology, Asan Medical Center, University of Ulsan, 388-1 PoongNap-Dong, SongPa-Gu, Seoul 138-736, Republic of Korea.
6 Supported by the Molecular Medical Research Group Program (00-J03-01-01-A-01).
References
- Received June 29, 2000.
- Copyright © American Society of Neuroradiology