Abstract
Summary: Mutations in the nuclear SURF1 gene are specifically associated with cytochrome c oxidase (COX)-deficient Leigh syndrome. MR imaging abnormalities in three children with this condition involved the subthalamic nuclei, medulla, inferior cerebellar peduncles, and substantia nigra in all cases. The dentate nuclei and central tegmental tracts were involved in two cases each (all instances), and the putamina, interpeduncular nucleus, and pallido-cortical-nigro-cortical tracts in one. MR imaging pattern recognition can suggest an underlying COX deficiency and should prompt investigators to search for SURF1 gene mutations.
Leigh syndrome (LS) is a progressive neurodegenerative disorder of infancy and childhood characterized by symmetric necrotic lesions with demyelination, vascular proliferation, and gliosis, involving the basal ganglia, diencephalon, brain stem, and spinal cord (1). Affected patients exhibit a variable clinical picture that frequently includes psychomotor retardation or regression, recurrent episodes of vomiting, failure to thrive, signs of brain stem and basal ganglia dysfunction, and lactic acidosis (2). LS is a biochemically and genetically heterogeneous disease that can be caused by defects of different enzymes involved in energy metabolism. An isolated, generalized defect of cytochrome c oxidase (COX), a component of the mitochondrial respiratory chain, is one of the most common biochemical abnormalities found in patients with LS (2, 3). Human COX is composed of 13 subunits, of which three are encoded by mitochondrial DNA (mtDNA) and the remainder by nuclear DNA. In addition, other nuclear genes, referred to as “COX assembly genes,” are required for its correct assembly and function (4). Loss-of-function mutations of one of these genes, SURF1, are specifically associated with COX-deficient LS (LS/COX−) (3). We report on the MR imaging findings in three children with LS/COX− harboring SURF1 gene mutations.
Case Reports
Clinical and neuroradiologic manifestations are summarized in the Table.
Clinical and MR findings
Case 1
This boy was born at term to healthy, nonconsanguineous parents after an uneventful pregnancy. The neonatal period was unremarkable. At 12 months of age, he was able to sit and roll around. At 14 months, his developmental milestones slowed and muscle hypotonia appeared. At 16 months, he had neurodevelopmental regression, generalized symmetrical hypotonia, and absent deep tendon reflexes. His weight, height, and head circumference were below the second centile.
Laboratory investigations revealed elevated blood lactate at rest (58 mg/dL; normal, 8–22 mg/dL) and increased lactate-pyruvate ratio (L/P=61.6; normal, 6–25). Electromyography (EMG) of the anterior tibialis muscle was suggestive of a neurogenic lesion. Motor nerve conduction velocity (MNCV) of the peroneal and median nerve was slowed. An electroencephalogram (EEG) showed mild nonspecific abnormalities. Histochemistry of skeletal muscle obtained by open biopsy showed virtually absent COX activity, and biochemical analysis of a muscle homogenate confirmed an isolated reduction of COX.
Common mtDNA mutations were ruled out by polymerase chain reaction-restriction fragment-length polymorphism screening, and the presence of large-scale deletions or duplications were excluded by Southern blot of the muscular mtDNA. By sequencing the entire coding region of the SURF1 gene, the patient was identified as compound heterozygous for a previously reported mutation at the splice-junction site of intron 3 (240 + 1G>T) and for a novel mutation (ie, a deletion involving 4 base pairs after nucleotide 530 [531_534delAAAT]) (5). Brain MR imaging was performed at age 16 months on a 0.5-T unit. It showed bilateral, symmetric areas of T2 prolongation involving the medulla above, at, and below the pyramidal decussation; T2 prolongation also involved symmetrically the inferior cerebellar peduncles, substantia nigra, central tegmental tracts, and subthalamic nuclei. The basal ganglia, thalami, and supratentorial white matter were spared. The patient experienced severe respiratory difficulties and died of cardiopulmonary arrest at 22 months of age. Autopsy was not performed.
Case 2
This girl is the first child of healthy, unrelated parents. She presented at age 12 months with failure to thrive, neurodevelopmental regression, and frequent vomiting. Physical examination showed facial dysmorphism, hirsutism, and generalized muscle hypotonia. Venous blood lactate was elevated at rest (32 mg/dL; normal, 8–22 mg/dL). Massive excretion of lactate and related metabolites was found, whereas plasma and urinary amino acids were normal. EMG showed pseudomyotonic discharges, whereas MNCV was normal. Histochemistry of skeletal muscle showed virtually absent COX activity and no ragged-red fibers. Biochemical analysis of a muscle homogenate confirmed an isolated COX reduction. Sequencing analysis of the SURF1 gene revealed a heterozygous single-base-pair deletion at nucleotide position 566 (566delG) in exon 6, which predicts a truncated surf1 protein, and a heterozygous mutation at the splice-junction site of intron 4. Brain MR imaging was performed on a 1.5-T unit when the patient was 15 months old. It showed T2 prolongation involving the medulla above, at, and below the pyramidal decussation and extending to the cervical spinal cord. Symmetric areas of T2 prolongation also involved the inferior cerebellar peduncles, dentate nuclei, substantia nigra, and subthalamic nuclei. The basal ganglia, thalami, and supratentorial white matter were unaffected. The child is alive at age 3 years.
Case 3
This boy was born at term to unrelated, healthy parents after an uneventful pregnancy. The neonatal period was unremarkable. During the first year of life, he showed failure to thrive and neurodevelopmental delay. At age 15 months, he was able to sit. During the second year of life, several episodes of palpebral ptosis and impaired consciousness occurred. Physical and neurologic examination showed facial dysmorphism, hirsutism, nystagmus, generalized muscle hypotonia, absent deep tendon reflexes, and breathing abnormalities. Laboratory investigations revealed elevated blood lactate at rest (42 mg/dL; normal, 8–22 mg/dL). EMG of the anterior tibialis muscle showed signs of both neurogenic and muscular involvement. MNCV of the peroneal and median nerve was slowed. EEG showed mild nonspecific abnormalities. Sural nerve biopsy showed mild demyelination. Histochemical and biochemical analysis of muscle biopsy showed severe reduction of COX activity. Sequencing analysis of the SURF1 gene revealed a homozygous mutation (772_773delCC) in exon 8. Brain MR imaging was performed at age 3 years on a 1.5-T unit (Fig 1). It showed diffuse T2 prolongation in the medulla, extending along the inferior cerebellar peduncles to both dentate nuclei. The midbrain also showed T2 prolongation involving several structures, such as the substantia nigra, central tegmental tract, interpeduncular nucleus, and pallido-cortical-nigro-cortical tracts. Both subthalamic nuclei showed symmetric T2 hyperintensity. The posteroinferior portions of the putamina also were hyperintense on T2-weighted images. The child is alive at age 4 years.

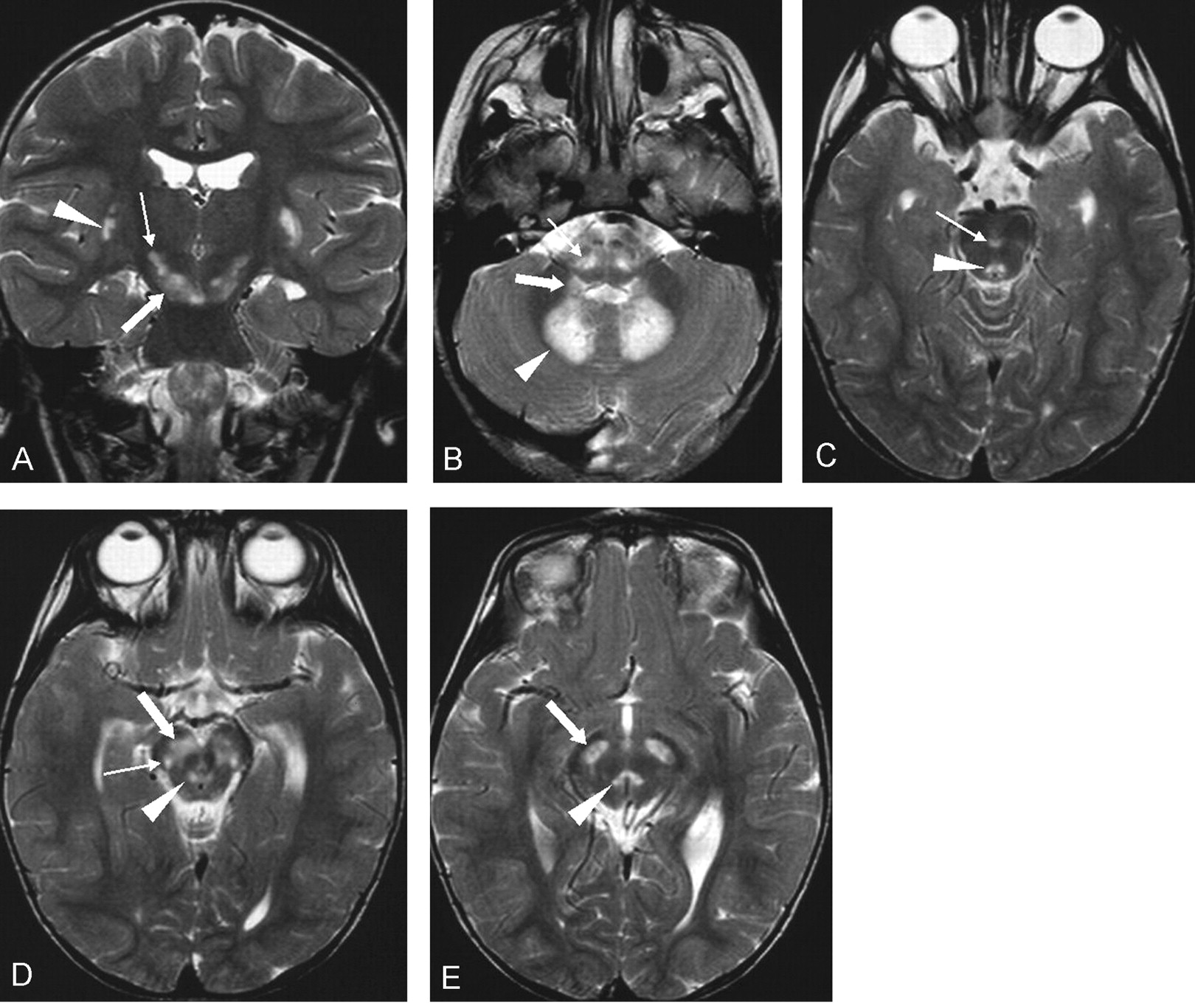{kind=link}
Case 3. MR imaging at age 3 years.
A, Coronal T2-weighted image (4500/120 [TR/TE]) shows symmetric T2 prolongation involving the subthalamic nuclei (thin arrow). The substantia nigra (thick arrow), posteroinferior portion of the putamina (arrowhead), and medulla are also involved.
B–E, Axial T2-weighted images (4500/120).
B, T2 prolongation involves the medulla (thin arrow), inferior cerebellar peduncles (thick arrow), and dentate nuclei (arrowhead).
C, Involvement of the interpeduncular nucleus (thin arrow) and central tegmental tract (arrowhead).
D, Diffuse involvement of the substantia nigra (thick arrow) is associated to discrete hyperintense lesions in the anatomic location of the central tegmental tract (arrowhead) and pallido-cortical/nigro-cortical tracts (thin arrow).
E, Simultaneous, symmetric involvement of the substantia nigra (thick arrow) and central tegmental tracts (arrowhead).
Discussion
Mitochondrial disorders are divided into somewhat ill-defined categories on the basis of their clinical, histologic, biochemical, and genetic features (6). LS represents a typical example of a clinical entity with blurred confines. Bilateral putaminal T2 prolongation has long been considered a consistent feature of LS on MR imaging studies (7, 8); however, the MR imaging picture may include involvement of the brain stem, basal ganglia, cerebral white matter, and sometimes the cerebral cortex in various combinations (6), and cases without putaminal abnormalities have been described (9). Such heterogeneity is probably related to the fact that LS is the end result of a number of different biochemical defects that affect many aspects of mitochondrial function (6). Unfortunately, correlations between MR imaging findings and corresponding biochemical and genetic defects have been rather limited so far.
LS/COX− with SURF1 gene mutations represents a relatively homogeneous clinical entity. Affected patients present at around 1 year of age with progressive encephalopathy, generalized hypotonia, trunk ataxia, oculomotor abnormalities, and central respiratory problems (3, 5). Although symmetric T2 prolongation involving the subthalamic nuclei has been considered as an almost distinctive mark of LS/COX− (10), MR imaging findings in patients with proved SURF1 gene mutations have only rarely been reported. Recently, Savoiardo et al (11) found symmetric T2 prolongation in the subthalamic nuclei and in the brain stem at different levels (medulla, pontine tegmentum, and periaqueductal area) in all their eight patients with LS/COX− harboring loss-of-function mutations of SURF1, with putaminal involvement in two of them. Conversely, they found basal ganglia involvement in 12 of 14 patients with SURF1-unrelated LS (including both COX deficiencies and other biochemical defects). Rahman et al (12) reported a SURF1 gene mutation case showing a supratentorial leukodystrophic pattern associated with T2 prolongation involving the medulla and dentate nuclei. Because as many as 31 different mutations of the SURF1 gene have been reported so far (5), one could expect to find some variability of the MR imaging findings among patients harboring different mutations. Indeed, “atypical” MR imaging findings, such as a leukodystrophic pattern, could represent examples of this variability.
Our results support the hypothesis made by Savoiardo et al (11) that bilateral involvement of the subthalamic nucleus and brain stem at various levels associated with mild or even absent basal ganglia abnormalities suggests the presence of COX deficiency and underlying SURF1 gene mutations. In our series, the subthalamic nuclei and brain stem were consistently abnormal, whereas the putamina were spared in two cases and only partially involved in one. The subthalamic nucleus is a lens-shaped structure located along the medial and cephalad margin of the peduncular portion of the internal capsule, whose caudal portion overlies the rostral portion of the substantia nigra. Involvement of the subthalamic nucleus is easily recognized on coronal T2-weighted MR images (Fig 1). Brain stem involvement is typically multilevel, and various structures can be abnormal in individual cases; however, certain features, such as the simultaneous involvement of the substantia nigra and central tegmental tract at level of the midbrain and T2 prolongation in the medulla, inferior cerebellar peduncles, and dentate nuclei were easily recognizable on axial T2-weighted images and appeared to be rather characteristic in our series (Fig 1). Other findings, such as the involvement of the interpeduncular nucleus and pallido-cortical-nigro-cortical tracts, to our knowledge represent a novel feature.
MR imaging pattern recognition has established itself as a powerful diagnostic method in several neurometabolic disorders. Recognition of the described MR imaging pattern in patients with LS should prompt a search for COX deficiency and SURF1 gene mutations; however, different mutations may result in different MR imaging phenotypes depending on which functional groups of the protein product are affected; therefore, the extent of MR imaging abnormalities in this disease has probably not yet been fully established. Although further studies on larger patient groups are required to determine the sensibility and specificity of MR imaging in the diagnosis of this condition, it is tempting to speculate that a positive MR imaging picture could directly lead to implement DNA testing, which is easily performed on leukocytes taken from peripheral blood, thereby avoiding muscle biopsy.
References
- Received July 15, 2002.
- Accepted after revision October 8, 2002.
- Accepted after revision October 8, 2002.
- Copyright © American Society of Neuroradiology