Abstract
BACKGROUND AND PURPOSE: The purpose of this study was to probe the cellular mechanism of healing in aneurysms after platinum coil embolization, by using multiple special stains and immunolabels.
METHODS: Elastase-induced aneurysms were created and embolized in 28 rabbits. Aneurysms were excised between 2 and 24 weeks after embolization. Specimens were embedded in paraffin, sectioned, and stained with hematoxylin-eosin, Masson trichrome, and multiple immunostains.
RESULTS: At 2 weeks, peripheral sparse spindle-nucleated cells were positive for α-smooth muscle actin (SMA), myosin, and vimentin, indicating myofibroblastic differentiation. At 4 weeks, all spindle-nucleated cells in the aneurysm were positive for SMA, myosin, desmin, and vimentin. Ten weeks after embolization, positive immunohistochemical staining in the cells populating the aneurysm significantly decreased. Mean positive SMA cells, per high-powered field were 5 ± 3, 45 ± 9, 10 ± 5, 0 ± 0, and 0 ± 0 at 2, 4, 10, 16, and 24 weeks, respectively. Findings of a Kruskal-Wallis test showed these data to be significantly different (P =.0001). Post hoc tests revealed significantly greater amounts of SMA-positive staining in the cells at 4 weeks compared with those at 2, 10, 16, and 24 weeks (P < .05). In addition, the 10-week group had significantly more positive cells than the 16- and 24-week groups (P < .05). There was a 78% decrease in apoptotic cells between 4 (37 ± 11) and 10 weeks (8 ± 4) after implantation. Apoptotic cells were completely absent beyond 10 weeks.
CONCLUSION: Aneurysm healing, in response to platinum coil embolization, appeared to progress through the stages of thrombus formation, granulated tissue organization, and loose connective tissue formation. Myofibroblasts, the key cellular component involved in healing, appeared within the aneurysm early. They progressively reduced in number with time and finally disappeared through the mechanism of apoptosis.
Although platinum microcoil therapy for aneurysms became routine clinical practice nearly a decade ago, the precise mechanism of healing for aneurysms after coiling remains poorly understood.1–8 Improved understanding of the healing mechanisms would be of vast clinical relevance because it might allow a more specific design for future-generation devices that could perform better than current ones.
Precise study of the healing mechanisms within coil-embolized aneurysms has been hampered by numerous factors. First, preclinical models, especially those using surgical anastomoses,9–16 likely do not reflect the biologic environment in human intracranial aneurysms. Second, the availability of coil-treated human aneurysm specimens has been extremely low. Last and perhaps most important, the tissue-processing techniques (methylmethacrylate) used for studying metal-bearing tissues have often yielded poorly stained samples and did not permit the utilization of advanced staining techniques such as immunohistochemistry17–25 and in situ labeling techniques.15,17–25 Without these, precise identification of cell types is difficult or impossible.
This study reports a longitudinal immunohistochemical examination of experimental aneurysms in rabbits. A new tissue-processing technique permitted implementation of a wide array of immunohistochemical stains. In addition, terminal deoxynucleotidyl transferase mediated nick and end-labeling (TUNEL), with in situ end-labeling of fragmented DNA, was used in some samples to identify the precise cellular mechanisms involved in healing of coiled aneurysms.
Materials and Methods
Aneurysm Creation
Elastase-induced saccular aneurysms were created in 28 New Zealand white rabbits (body weight, 3–4 kg) by using the rabbit elastase model. The Institutional Animal Care and Use Committee approved all procedures before initiation of the study. This was a retrospective study, and the tissue from these aneurysms was used in other experiments before being used in this study. Detailed procedures for aneurysm creation have been described in depth elsewhere.26 Briefly, anesthesia was induced with an intramuscular injection of ketamine, xylazine, and acepromazine (75, 5, and 1 mg/kg, respectively). Using sterile technique, we exposed the right common carotid artery (RCCA) and ligated it distally. A 1- to 2-mm beveled arteriotomy was made, and a 5F vascular sheath was advanced retrograde in the RCCA to a point approximately 3 cm cephalad to the origin of RCCA. A 3F Fogarty balloon was advanced through the sheath to the level of the origin of the RCCA with fluoroscopic guidance and was inflated with iodinated contrast material. Porcine elastase (Worthington Biochemical, Lakewood, NJ) was incubated within the lumen of the common carotid artery above the inflated balloon for 20 minutes, after which the catheter, balloon, and sheath were removed. The RCCA was ligated below the sheath entry site, and the incision was closed.
Aneurysm Embolization
Aneurysms were permitted to mature for at least 21 days after creation. The anesthesia described in the aneurysm creation was used for aneurysm embolization. Using sterile technique, we performed surgical exposure of the right common femoral artery. The artery was ligated distally by using 4-0 silk suture, and a 22-gauge angiocatheter was advanced retrograde into the artery. A guidewire (0.018 in) was passed through the angiocatheter followed by placement of a 5F vascular sheath. Heparin (100 U/kg) was administered intravenously. A 5F catheter was advanced into the brachiocephalic artery. Using coaxial technique, with a continuous heparinized saline flush, we advanced a microcatheter into the aneurysm cavity. The size of the aneurysm cavity was assessed by using direct comparison to radiopaque sizing devices during digital subtraction angiography (DSA). Aneurysms were embolized with platinum coils as previously described.22 Aneurysm cavities were densely packed in all cases. After the embolization, a final control DSA was performed. The catheters and sheath were removed, the femoral artery was ligated, and the incision was closed.
Tissue Harvest
At the time of sacrifice, the subjects were deeply anesthetized as described previously and then were sacrificed by using a lethal injection of pentobarbital. The chest cavity was opened, and the mediastinum was dissected. Saline and formalin were rapidly injected to flush the vessels through the aortic arch. The proximal great vessels, including the coil-embolized segment of artery, were exposed and dissected free from surrounding tissues. Aneurysms were harvested at 2 weeks (n = 5), 4 weeks (n = 6), 10 weeks (n = 5), 16 weeks (n = 6), and 24 weeks (n = 6). Specimens were removed and were immediately fixed in 10% neutral buffered formalin.
Histologic Processing
The fixed tissues were dehydrated in an ascending series of ethanol, cleared in xylene, and embedded in paraffin. Using an IsoMet Low Speed Saw (Buehler, Lake Bluff, Ill) with a Series 15HC Diamond Blade and Isocut Fluid (Buehler), we sectioned the aneurysms at 1000-μm intervals through the portion bearing metallic coils in a coronal orientation. Under a dissection microscope, the metallic coil fragments were carefully removed. After removal of all coil fragments, the sections were re-embedded in paraffin blocks. These blocks were sectioned using a microtome with disposable blades at 5- to 6-μm intervals. Sections were floated on a water-bath at 42°C to remove the wrinkles, then mounted on glass slides, and allowed to dry overnight in an oven at 37°C.
This retrospective study used tissue specimens from other studies. The size of each aneurysm was finite. Therefore, some tissue specimens were processed for all 4 techniques, including TUNEL, whereas others had to be limited to hematoxylin-eosin (H&E), Masson trichrome, and immunohistochemistry staining (Table 1).
Distribution of subjects for tissue processing
Staining
H&E
At least 2 sections from each block were stained with H&E for conventional histopathologic evaluation.
Masson Trichrome Stain
Serial sections were stained with Masson trichrome. The slides were deparaffinized and hydrated in distilled water, then were placed in Bouin’s solution for 30 minutes in a 56°C water bath. They were rinsed in running tap water until the sections were colorless, stained in filtered Weigert’s iron hematoxylin working solution for 10 minutes, and then washed in tap water for 5 minutes. After being rinsed in distilled water, they were stained in Biebrich scarlet-acid fuchsin for 4 minutes and then rinsed in distilled water. They were placed in phosphomolybdic-phosphotungstic acid working solution for 12 minutes and then placed in aniline blue solution for 20 minutes. Finally, after the slides were rinsed in distilled water, they were placed in 1% acetic acid for 4 minutes, then dehydrated through alcohols, cleared in xylene, and mounted with Shandon EZ mount (Thermo Electron, Pittsburgh, Pa).
Immunohistochemistry
The VECTASTAIN Elite ABC system (Vector Laboratories, Burlingame, Calif) was used for single-label immunohistochemistry.27 Sections were deparaffinized and incubated with 0.3% hydrogen peroxide at 37°C for 20 minutes and then pretreated with 0.1 mol/L of citric acid buffer in a microwave for 15 minutes. Slides were left to cool at room temperature for 30 minutes before being rinsed in phosphate buffered saline (PBS). Sections were then incubated with normal 5% horse serum for 20 minutes at 37°C, followed by incubation with the appropriate primary antibody for 1 hour. The primary antibodies used included a monoclonal mouse anti-human smooth muscle actin (SMA, 1:200, Dako Denmark A/S, Glostrup, Denmark), a mouse monoclonal antimyosin (myosin heavy chain, clone HSM-V 1:1000, Sigma-Aldrich, St. Louis, Mo), a mouse monoclonal antidesmin (Clone DE-U-10, 1:150, Sigma-Aldrich), and a mouse monoclonal antivimentin (Clone LN-6, 1:200, Sigma-Aldrich). The slides were rinsed in PBS and incubated with biotinylated secondary Horse Antimouse IgG (Vector Laboratories) at 37°C for 1 hour. After the PBS rinse, the sections were incubated with VECTASTAIN Elite ABC reagent (Vector laboratories) at 37°C for 45 minutes. Finally, the sections were stained with DAB (diaminobenzidine tetrahydrochloride) Substrate Kit (Vector Laboratories) for horseradish peroxidase (HRP) followed by counter stain with Gill hematoxylin. Positive controls included sections of human tissues that were positive for SMA, myosin, desmin, and vimentin. Negative controls were performed with nonimmune normal serum used instead of the primary antibody.
Specific Labeling of Nuclear DNA Fragmentation
TUNEL28 was performed on the sections of 13 coiled aneurysms at 4 weeks (n = 4), 10 weeks (n = 3), 16 weeks (n = 1), and 24 weeks (n = 5) for specific labeling of nuclear DNA fragmentation with DeadEnd Colorimetric TUNEL System (Promega Corp, Madison, Wis). The sections were deparaffinized and rehydrated as described previously, followed by fixing them in 4% paraformaldehyde for 15 minutes. They were incubated with 20 μg/mL of proteinase K for 20 minutes at room temperature, and then sections were rinsed with PBS and kept in the equilibration buffer for 10 minutes at room temperature. They were incubated with 100 μL of working terminal deoxynucleotidyltransferase (TdT) reaction mix (containing recombinant terminal deoxynucleotidyltransferase [rTdT] enzyme) in a humid atmosphere for 1 hour at 37°C. The reaction was terminated by immersing the slides in water for 15 minutes. The sections were then immersed in 0.3% hydrogen peroxide for 5 minutes at room temperature to inactivate endogenous peroxidase and then were rinsed with PBS and incubated with 100 μL of streptavidin for 30 minutes at room temperature. Then they were rinsed with PBS, and the sections were stained with DAB. Positive controls consisted of incubating sections with DNase i. A negative control was performed by incubating the section with rTdT reaction mix without the rTdT enzyme.
Two experienced observers independently reviewed all the slides by using an Olympus BH2 microscope (Olympus America, Melville, NY). Images were recorded with a Spot RT Digital Camera (version 3.0, Diagnostic Instruments, Sterling Heights, Minn). In early time point samples and some late samples, unorganized thrombus or fibrin occupied at least part of aneurysm dome and neck; coils also occupied space in these areas. Thus, for the purpose of quantifying cells, 5 random high-powered organized areas in both the aneurysm cavity and neck area were imaged. Both the number of positive SMA-stained cells and TUNEL-stained cells were recorded. Both the SMA and TUNEL counts for each specimen are shown as mean ± standard deviation.
Statistical Analysis
Kruskal-Wallis tests were used to compare the number of SMA-positive cells between weeks, whereas a χ2 test was used for apoptotic cells. Any differences found with the overall Kruskal-Wallis test were further elucidated by using a Tukey post hoc test. A value of P < .05 was considered significant.
Results
Conventional Staining
The 5 samples harvested at 2 weeks after coil implantation had unorganized thrombus that filled most of the aneurysm cavity. This was associated with scattered, sparse, spindle- or satellite-like cells at the periphery. Two of 5 samples had unorganized thrombus traversing the aneurysm neck. There was no evidence of endothelial cells along the necks. The other 3 samples had unorganized thrombus with a partial endothelial cell lining across their necks. Collagen deposition was absent in the dome and at the neck in all 5 specimens.
Four weeks after coil embolization, 2 of the 6 samples had aneurysm lumens that were mostly filled with loose connective tissue, consisting of spindle cells and thin-walled vessels. In the center of the loose tissue were associated, small areas of poorly organized thrombus. Three other specimens at 4 weeks had aneurysm domes with large areas of unorganized thrombus accompanied by loose connective tissue at the periphery. These 5 aneurysms had unorganized fibrin and/or laminated mixed thrombus across the neck. “Laminated mixed thrombus ” is a term denoting alternating layers of platelet aggregates and a network of fibrin, which entrap red and white blood cells. A single specimen displayed completely organized loose connective tissue and a thin layer of fibrous tissue traversing the entire neck. This aneurysm was the smallest in this series, with a neck of only 1.8 mm in diameter. Four of these 6 samples had chronic inflammatory foci surrounding/between the coil winds. Masson trichrome staining revealed sparse, disorganized collagenous fiber deposition among spindle cells in 1 sample. Collagen deposition was not present in the other 5 aneurysms at this time point.
Two of 5 aneurysms displayed large areas of unorganized or poorly organized thrombus associated with peripheral loose tissue, which filled the aneurysm dome and traversed the neck 10 weeks after embolization. Three specimens had vascularized, loose, hypocellular connective tissue filling the aneurysm lumen. Small areas of poorly organized thrombus were also present in at least a portion of 2 of these 3 samples. Localized, chronic inflammatory cell infiltration was observed in 2 of 5 subjects at this time point. Five samples had thin layers of fibrinous or fibrous tissue across the neck. There was no collagen deposition identified by Masson trichrome staining.
Sixteen weeks after embolization, all 6 aneurysm lumens were filled with a loose, hypocellular meshwork tissue and thin-walled neovessels, accompanied by a thin layer of fibrous or fibrinous tissue across the neck. A single sample possessed a small area of poorly organized thrombus within the loose tissue. Three specimens had marked localized chronic inflammatory cell infiltration primarily consisting of lymphocytes surrounding some coil winds. Mild chronic inflammatory infiltration was observed in the other 3 samples. In a single specimen, multiple giant cells were noted within some coil winds near the aneurysm wall.
All 6 aneurysm lumens were completely filled with loose hypocellular connective tissue 24 weeks after embolization. Five necks were covered with a thin layer of fibrous tissue. The sixth neck was composed of unorganized fibrin. One sample displayed marked chronic inflammatory cell infiltration surrounding some coil winds. Collagen deposition was absent on Masson trichrome staining in all samples 16 and 24 weeks after embolization.
Immunohistochemistry
Two weeks after embolization, the sparse spindle- or satellite-like cells at the periphery displayed a strong positive reaction with SMA and a weak reaction with myosin heavy chain and vimentin, indicating differentiated myofibroblasts. Differentiated myofibroblasts are specialized contractile fibroblasts that are common during wound-healing. These myofibroblasts have features in common with smooth-muscle cells. They are typically positive for vimentin and SMA; they also express varying combinations of vimentin, smooth-muscle actin, desmin, and smooth-muscle heavy-chain myosin.
In all 6 samples at 4 weeks, the spindle-nucleated cells within the aneurysm dome, in addition to the cells lining the walls of the neovessels, showed a strong immunoresponse to SMA (Fig 1A). These SMA-positive cells displayed a moderately positive reaction with antibodies for myosin, desmin, and vimentin, indicating a myofibroblastic immunophenotype.

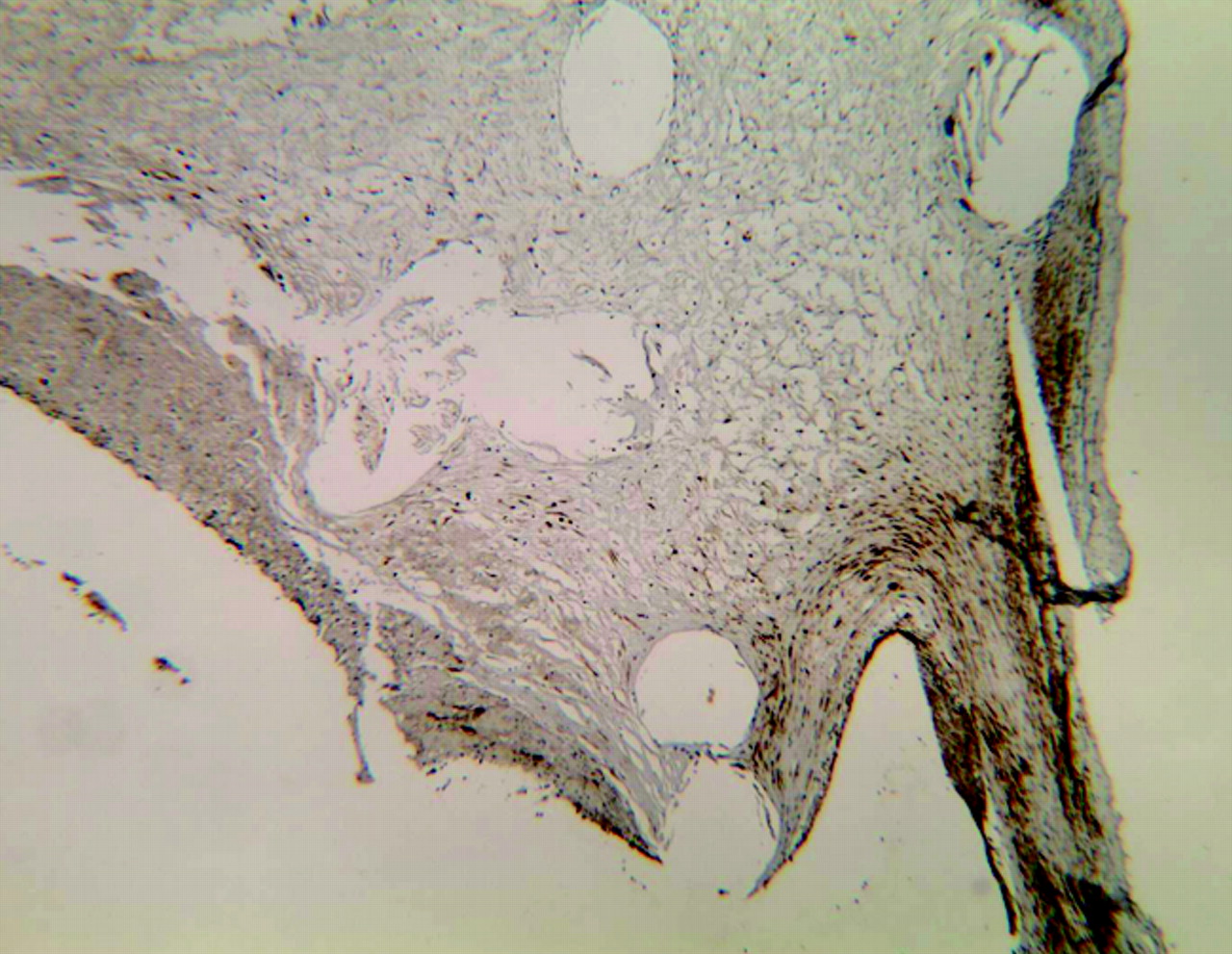
Photomicrographs of rabbit aneurysms embolized with platinum coils show that spindle cells within the aneurysm dome at 4 weeks (A) are positive for SMA and negative for SMA at 16 weeks (B) (immunohistochemistry, antibody to SMA, original magnification ×200).
At 10 weeks, most cells embedded in the loose matrix in all 5 samples were negative for SMA, myosin, desmin, and vimentin. There were, however, some sparse spindle cells, as well as the cells lining the wall of neovessels, that stained positive for SMA and vimentin (Fig 2). However, the attenuation and extent of staining was markedly diminished compared with that seen at 4 weeks.

Photomicrograph of a rabbit aneurysm 10 weeks after embolization with platinum coils shows sparse cells within the dome that are positive for SMA staining (arrow); no positive cells were observed at the neck (dashed arrow). The cells at the side edge of neck (the transition zone of aneurysm wall to aneurysm neck) are positive for SMA (dotted arrow) (immunohistochemistry, antibody to SMA, original magnification ×60).
At 16 and 24 weeks after embolization, all the cells embedded in the loose matrix of aneurysm cavity were negative for SMA, myosin, desmin, and vimentin (Fig 1B).
The mean numbers of cells per high-powered field with positive staining for SMA at 2, 4, 10, 16, and 24 weeks after embolization were 5 ± 3, 45 ± 9, 10 ± 5, 0 ± 0, and 0 ± 0, respectively. The Kruskal-Wallis test showed a significant overall difference (P = .0001). Tukey post hoc tests between individual groups revealed a significantly greater amount of SMA-positive staining in the cells at 4 weeks compared with 2, 10, 16, and 24 weeks (P < .05). In addition, the 10-week group had significantly more positive cells than the 16- and 24-week groups (Table 2). These data indicated that myofibroblasts, abundantly present at 4 weeks, gradually disappeared with time. The remaining cells were considered consistent with fibrocytes, without contractile and matrix-producing ability.
Summary of histologic findings in each group
TUNEL
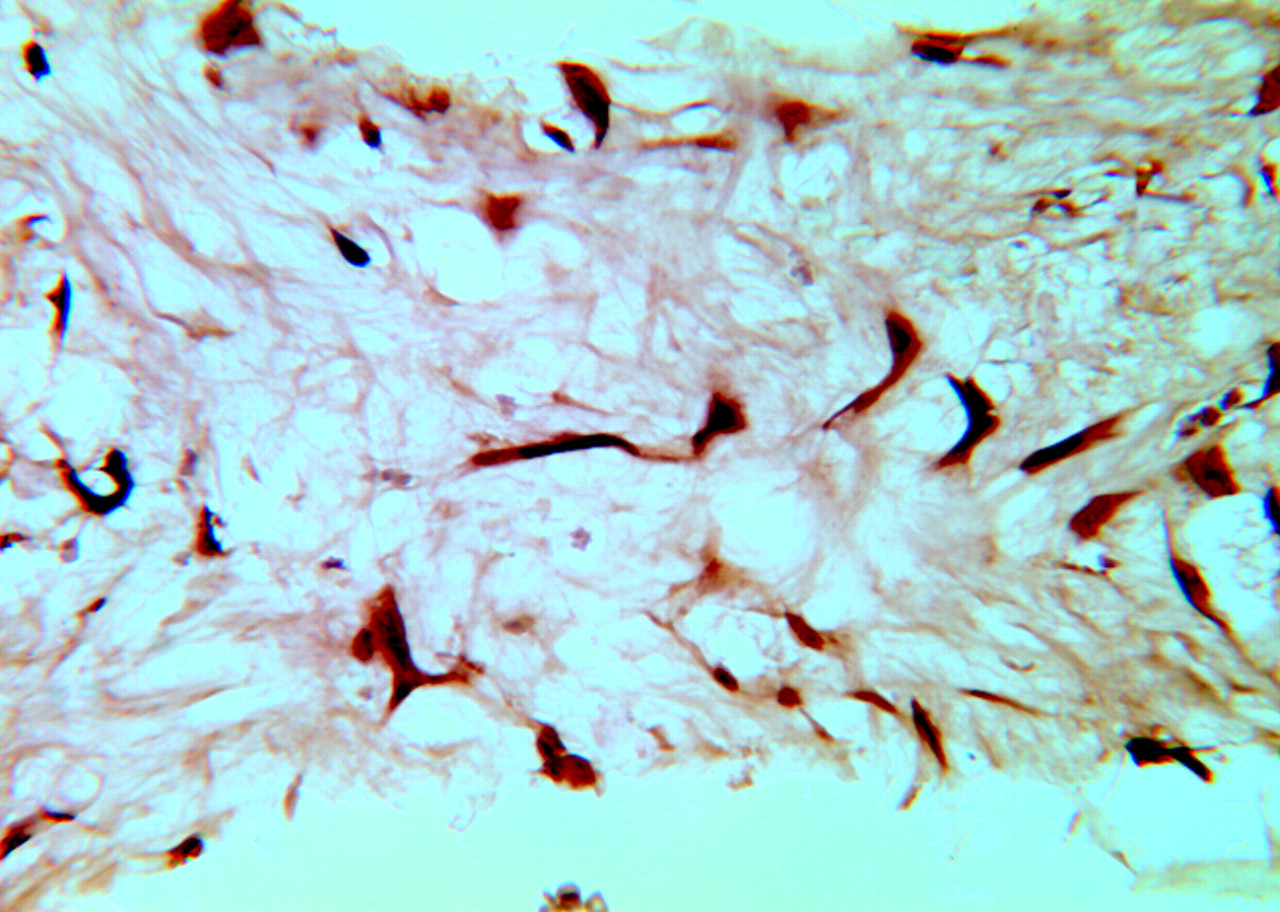
The TUNEL assay was not performed on the 2-week samples because there were almost no nucleated cells in the specimens. Serial sections of 4 samples at 4 weeks were stained for apoptosis analysis. Most of the spindle-nucleated cells (myofibroblasts) in the aneurysm dome were TUNEL-positive, indicating apoptotic change (Fig 3) for all specimens. Ten weeks after embolization, only scattered sparse TUNEL-positive cells were detected. No TUNEL-positive cells were observed in any specimen 16 and 24 weeks after embolization.

{kind=link}
{kind=link}
{kind=link}
Photomicrograph of TUNEL staining for a 4-week rabbit aneurysm embolized with platinum coils. The attenuated brown or dark brown signal intensity is localized within the nuclei of the spindle cells, indicating positive staining (TUNEL, original magnification ×200).
The mean number of TUNEL-positive apoptotic cells at 4, 10, 16, and 24 weeks after embolization were 37 ± 11, 8 ± 4, 0 ± 0, and 0 ± 0, respectively. Sixteen-week data (n = 1) were dropped out of the analysis because of the small sample size. A χ2 test was conducted on the remaining aneurysms on the basis of the presence or absence of TUNEL-positive cells. The χ2 test showed that there was a significant difference (P =.0025) between the 3 time points (4, 10, and 24 weeks). There was a 78% decrease in apoptotic cells between 4 and 10 weeks. Apoptotic cells were completely absent beyond 10 weeks. The results for all time points are summarized in Table 2.
Discussion
As early as 1913, Anitschkow29 used cholesterol-fed rabbits as a model to study atherosclerosis. More recently, rabbit models have been used to study such disparate vascular injuries as liposome-mediated transfection of endothelial nitric oxide synthase and transplant arteriopathy,30 MMP1 and restenosis,31 vein graft stent placement,32 and low-molecular-weight heparin and restenosis,33 to name a few. However, there are no studies, to our knowledge, that used any advanced in situ labeling techniques such as immunohistochemistry and TUNEL to elucidate, in detail, the cellular mechanisms involved in the healing of coil-embolized aneurysms in a rabbit model.
Multiple immunohistochemical markers were used to identify the cell types in the aneurysms. Early after coil implantation, the dome comprised primarily unorganized thrombus. Within this thrombus, centripetal infiltration of myofibroblasts was observed. This infiltration may have originated from the vessel wall or may have represented transformation of blood elements present in the thrombus. Within 4 weeks, infiltration of the dome with myofibroblasts was complete, with near-total resolution of thrombus. Deposition of structural proteins such as collagen remained absent, and tissue coverage at the neck was thin at all time points.
The key cells present in this study appeared large, spindle-shaped, and often stellate (spiderlike), having long cytoplasmic extensions. These cells possessed distinct acidophilic-to-amphophilic fibrillar cytoplasm with cablelike stress fibers. The nuclei were often indented, containing nucleoli and fine granular chromatin. Immunohistochemistry showed that these cells were always positive for SMA and vimentin and sometimes were combined with desmin or myosin expression. On the basis of these features, these cells were designated as myofibroblasts, which stain strongly for SMA and represent the primary cell type within the aneurysm dome at 4 weeks. In the chronic phase (>16 weeks), the expression of markers for myofibroblasts essentially vanished in the dome.
These findings can be placed into perspective by review of other related literature, including wound-healing studies and thrombus-organization studies. Thrombus organization has been studied in the rat aorta.34, 35 Early infiltration with neutrophils is largely replaced with monocyte infiltration within 1 week. Spindle cells expressing SMA become prominent at 1 or 2 weeks and remain for up to 10 months. Collagen is also present within experimental thrombus in rats. The data from the present experiments share some similarities with prior studies on thrombus organization. The exceptions are the observation that SMA-positive spindle cells in aneurysms essentially disappear by 3 months and that collagen is all but absent in all specimens.
Myofibroblasts play a prominent role in wound-healing; thus, they may offer some insight into the healing process after coil embolization of elastase-induced aneurysms in rabbits. In the wound-healing setting, myofibroblasts are thought to originate from cells termed “proto-myofibroblasts,”36 which are present within the dermis or other tissues. In response to stimuli, including mechanical tension as well as exposure to factors such as transforming growth factor-β (TGF-β),37, 38 proto-myofibroblasts express contractile proteins and become myofibroblasts. When contraction stops and the wound is fully epithelialized, myofibroblasts progressively disappear,39 either through the differentiation of myofibroblasts into the quiescent form (fibrocytes) or selectively through programmed cell death.39–41
Programmed cell death, or apoptosis, is the process whereby cells are induced to activate their own death or cell suicide. Identification of apoptosis in tissue sections has been facilitated by specific immunolabeling of nuclear DNA fragmentation with terminal TdT.28 Using this method, along with immunohistochemistry, illustrated the fact that myofibroblast differentiation achieves its peak at 4 weeks after coil embolization, and this differentiation ended with the cell death at the same 4-week time point. The observed findings in this rabbit model coincided with the timing observed in wound-healing, suggesting close homology between wound-healing and aneurysm-healing.
These observations lend insight into the mechanism of action for healing in aneurysms embolized with platinum coils and, perhaps more importantly, may lead to more specifically designed modified devices. First, these findings suggest that substantial changes in the cellular environment occur with time. Cells that might effect healing, including the myofibroblast, become apparent weeks after treatment and then essentially disappear. In light of the time-dependent changes, any design of a modified device with an active agent should require that the agent remain present for at least several weeks after implantation. These data also suggest that a cell considered relevant for healing, the myofibroblast, is present in high concentrations within experimental aneurysms, but these same myofibroblasts are not evident chronically. Modified devices aimed at sustaining the persistence and expression of contractile cells by using growth factors such as TGF-β, for example, may be of benefit. Finally, the amount of collagen within aneurysms is minimal, unlike that found in organized thrombus in other locations, and may offer a separate target for modified devices.
This study has several limitations. First, the model used is not that of an intracranial aneurysm. Thus, significant differences in the cellularity of the vessel wall between the rabbit model and human intracranial aneurysms likely exist. In addition, though cell types have been identified in experimental aneurysms, the exact source of the cells remains unknown. The myofibroblasts may migrate from the vessel wall or may represent transformation of blood elements. This model cannot differentiate between these 2 sources of cells. In addition, this was a retrospective study. Limited amounts of tissue did not permit TUNEL staining for all samples. In addition, only 5 time points were chosen for observation. We have observed that healing started at 2 weeks and the healing process tended to plateau at 12–18 weeks in previous studies of the rabbit aneurysm model. Assuming that the healing process occurred in a similar manner, we chose these time points for the study. Although these data show that myofibroblasts disappeared with time because of apoptosis, the exact nature of the stimulus leading to apoptosis during the aneurysm-healing after coil embolization, in this model, remains to be answered. In addition, the apoptotic mechanism described here in the rabbit model also needs to be confirmed in the human brain aneurysm. Finally, elastase-induced aneurysms in the rabbit model are relatively small and do not often undergo the degree of coil compaction seen clinically.
Conclusion
Responses of rabbit elastase–induced aneurysms to platinum coil embolization appeared to progress through thrombus formation, granulated tissue organization and, finally, loose connective tissue formation. Myofibroblasts, the key cellular component involved the healing, developed early, progressively reduced with time, and finally disappeared through apoptosis.
These findings may permit a more specific design of modified coils aimed at improving aneurysm healing.
Footnotes
This work was supported by research grant NS42646 from the National Institutes of Health.
Presented at the 43rd annual meeting of the American Society of Neuroradiology, Toronto, Ont, Canada, April–May 2005.
References
- Received August 21, 2005.
- Accepted after revision September 14, 2005.
- Copyright © American Society of Neuroradiology