
Abstract
Summary: We report the MR and clinical findings of two patients with growth hormone deficiency and posterior pituitary ectopia (PPE). Possible causes of PPE are discussed.
Congenital pituitary dwarfism caused by idiopathic deficiency of growth hormone (CIGHD) accounts for approximately 10% of all cases of dwarfism (1). The underlying cause for this entity is unclear in most cases. In recent years, several MR studies have shown hypothalamic-hypophyseal abnormalities in these patients (2, 3). These abnormalities consist of hypoplasia of the anterior pituitary gland, absence or severe hypoplasia of the pituitary stalk, and a suprasellar hyperintensive spot on T1-weighted images: the so-called posterior pituitary ectopia (PPE) (4). Two hypothetical causes of PPE are discussed in the literature: 1) a transection of the pituitary stalk during a traumatic birth or an insult of the stalk during perinatal asphyxia (2, 3, 5); 2) an embryonic abnormality/defect in pituitary gland organogenesis (4, 6, 7). We report two brothers with CIGHD and imaging features of PPE supporting the organogenetic theory.
Case Reports
Case 1
An 8-year-old boy was referred to our pediatric endocrinology unit for growth retardation. Medical history revealed that he underwent normal, headfirst delivery at birth. His birth weight (3300 g) and birth length (55 cm) were normal. There were no pre-, peri-, or postnatal complications. Except for the retarded growth, there were no anamnestic abnormalities during childhood nor was there evidence of chronic disease. Prior head injury, visual disturbance, and history of diabetes insipidus were denied.
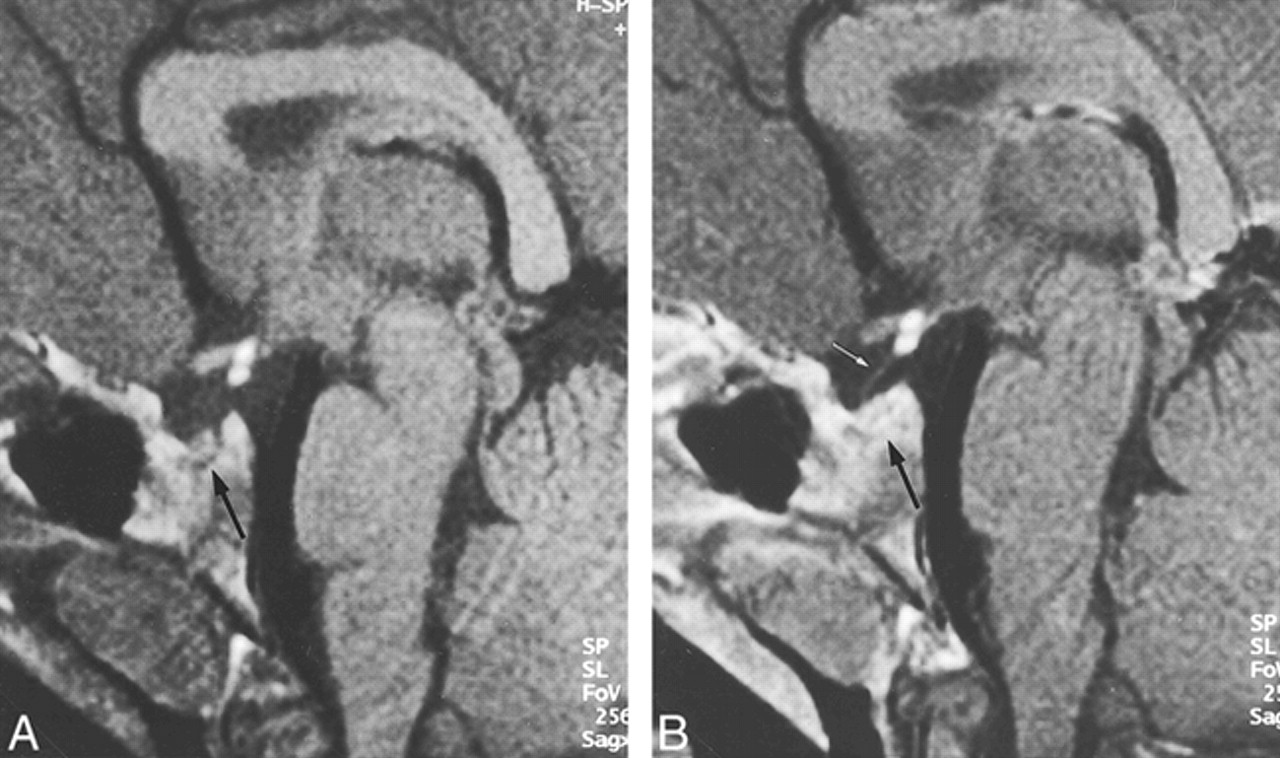
Physical examination revealed a body length of 114.8 cm, which is 10 cm below the 3rd percentile. Weight (19.7 kg), sitting height (63 cm), and arm range (118 cm) corresponded to body length. External genitalia and pubic hair were normal for his age. A radiograph of the left hand showed a bone age of 6 years. Laboratory and hormonal studies showed thyroid hormones, cortisol, erythrocyte sedimentation rate (ESR), hemoglobin A1c, hepatic enzymes, blood count, and renal function were normal. Low insulinlike-growth-factor-1-binding-protein-3 (IGF1-BP3) (1.3 mg/L; normal > 1.35 mg/L) and insulinlike-growth-factor-1 (IGF1) (51 ng/mL; normal > 126 ng/L) were compatible with growth hormone deficiency (GHD). The Insulin-Arginin-Test and Growth Hormone Releasing Factor (GRF)-Test were normal, with a maximum increase of growth hormone (GH) to 18.2ng/mL and 13.3 ng/mL. The spontaneous secretion of GH during the night was decreased with two peaks of 3.7 and 7.1 ng/mL. These results are compatible with neurosecretory dysfunction, consisting of a disorder of endogenous GH release that does not affect the ability to produce GH after exogenous stimulation. Single-strand conformation polymorphism (SSCP) of all exons of the Pit-1 and Prop-1 genes revealed no mutations. MR imaging of the brain and pituitary was performed on a 1.5-T system (ACS NT; Philips, Best, the Netherlands). Sagittal and coronal T1-weighted images (450/20 [TR/TE]; slice thickness, 2 mm; number of signals averaged (NSA), 2; field of view (FOV), 200; matrix, 205 × 256) were obtained through the sella region before and after IV application of Gd-DTPA (Magnevist; Schering, Berlin, Germany). The MR study revealed a suprasellar bright spot of 4.5-mm diameter, consistent with ectopic posterior pituitary tissue. In addition, a small pituitary stalk and a hypoplastic anterior pituitary gland were present (Fig 1). The other cerebral structures were unremarkable.

PPE in an 8-year-old boy depicted by T1-weighted (450/20) sagittal (A) and coronal (B) MR images. PPE is represented by a suprasellar bright spot of 4.5-mm diameter. Note the hypoplastic pituitary stalk (small arrow) and hypoplastic anterior pituitary gland (large arrows)
Case 2
The brother of case 1, a 5-year-old boy, was also referred to our pediatric endocrinology unit for growth retardation. The patient also underwent normal headfirst delivery at birth. His birth weight (3540 g) and birth length (52 cm) were normal. There were no pre-, peri-, or postnatal complications. Vegetative anamnesis was normal. Chronic illnesses, head injuries, visual disturbances, and symptoms of diabetes insipidus were denied. No pharmacological therapy was initiated.
Physical examination revealed a body length of 88.9 cm, which is 12 cm below the 3rd percentile. His body weight (10.8 kg), sitting height (53.2 cm), and arm range (89.1 cm) were consistent with the body length. External genitalia and pubic hair were normal for his age. A radiograph of the left hand showed a bone age of 1.5 years.
Laboratory and hormonal studies showed thyroid hormones, cortisol, ESR, HbA1c, ferments, blood count, and renal function were normal. Low IGF1-BP3 (0.9 mg/L; normal > 1.2 mg/L) and IGF1 (9ng/mL; normal > 24 ng/L) were compatible with GHD. The Insulin-Arginin-Test and GRF-Test showed a maximum increase of 6.3 ng/mL and 9.3 ng/mL. These results were compatible with a partial GH deficiency. Based on these findings, the spontaneous secretion of GH was not evaluated. SSCP of all exons of the Pit-1 and Prop-1 genes revealed no mutations.
MR images of the brain and pituitary were acquired on a 1.5-T System (Magnetom Vision; Siemens, Erlangen, Germany). Sagittal and coronal T1-weighted images (400/14 [TR/TE]; slice thickness, 3 mm; NSA, 3; FOV, 158 × 180; matrix, 205× 256) were obtained through the sella region before and after IV application of Gd-DTPA. The MR study revealed two suprasellar hyperintense spots of 2.5-mm diameter each, consistent with two parts of an ectopic posterior pituitary gland. The anterior pituitary gland was hypoplastic. The pituitary stalk was hypoplastic and only visible after IV contrast medium administration (Fig 2). The other cerebral structures were unremarkable.

PPE in a 5-year-old boy depicted by T1-weighted (400/14) sagittal MR images before (A) and after (B) contrast medium administration (courtesy of Dr. U. Müller-Lung, Cologne). PPE is represented by two suprasellar bright spots of 2.5-mm diameter each. Note the hypoplastic, enhancing anterior pituitary gland (large arrows). The hypoplastic pituitary stalk is only visible after contrast medium application (small arrow)
Clinical and MR Examination of the Patients' Parents
Because a hereditary cause for the presence of CIGHD was suspected in the two brothers, the parents were examined clinically and by MR imaging. The father showed a lower-than-average height of 162 cm. The mother, with a height of 162 cm, was still within normal range. Both parents had unremarkable pubertal development and had normal clinical examinations. MR imaging of both parents showed no abnormalities.
Discussion
The pituitary gland consists of two different developmental parts. The adenohypophysis or anterior pituitary is considered ectodermal in origin and develops from an evagination of the stomodeum (Rathke-cleft). The neurohypophysis or posterior pituitary is of neuroectodermal origin and develops as a downward extension of the diencephalon (Infundibulum). Hypothalamic releasing hormones reach the anterior pituitary via the infundibular portal system. Antidiuretic hormone and oxytocine are transported to the posterior pituitary via neurosecretory cells travelling through the infundibular stalk. On MR images, the anterior and posterior pituitary and infundibular stalk are usually well delineated. On T1-weighted images, the posterior pituitary shows hyperintense signal. The anterior pituitary gland is of intermediate signal intensity.
The cause of the hyperintense signal of the posterior pituitary on T1-weighted images is controversial. One author suggested the source of the signal to be neurosecretory granules (8). Another report states that the high signal intensity might be related to lipid in the posterior lobe pituicytes (9).
GHD comprises a spectrum of disorders of varying pathogeneses and pathologic characteristics and can be classified as isolated GHD or part of multiple pituitary hormone deficiencies (MPHD). Rare causes for GHD are inflammation, tumor, or trauma of the sellar region. Most of the cases are sporadic and still labeled idiopathic, although some cases with GH gene deletions have been described. Morphologic changes have been observed on MR images in a high percentage of pituitary dwarfism as well as in panhypopituitarism. These changes consist of a PPE at the median eminence of the hypothalamus, absent or hypoplastic infundibular stalk, and facultative hypoplastic anterior pituitary gland. A disturbed communication between the hypothalamus and the pituitary gland can be considered responsible for the development of PPE, and different theories have developed as to the underlying etiology. An increased incidence of breech delivery and birth asphyxia among newborns who later developed pituitary dwarfism has led to the suggestion that stalk ischemia or transection is the cause of PPE (2, 3, 5, 10). Nonetheless, it appears unlikely that a trauma severe enough to dissect the infundibular stalk should not lead to any further associated brain damage.
Other authors have found evidence that PPE is part of a congenital defect. This hypothesis is supported by a large number of cases of PPE without birth complications and the association of PPE with other brain anomalies that cannot be explained by perinatal asphyxia (4, 6, 7). Triulzi reported a total of 12% associated brain anomalies in 59 cases of PPE, including optic chiasma hypoplasia, partial dysgenesis or agenesis of septum pellucidum, absence of internal carotid artery and carotid canal, platybasia, and basilar impression (4). In the two reported cases, no such abnormality was observed.
The genetic theory is also supported by the findings among patients with MPHD that revealed mutations in the Pit-1 gene and the Prop-1 gene, necessary for the specification of pituitary cells (11). Although a genetic analysis did not reveal any mutations in the above-mentioned Pit-1 gene and Prop-1 gene, a hereditary etiology in our patients can still be assumed. This is because disturbances in the development of the pituitary gland and infundibular stalk most likely are a multifactorial event, and other genes not yet identified may play an important role in the proper specification of pituicytes. The normal birth of both patients without breech presentation, perinatal asphyxia, or other birth complications is evidence against any traumatic or ischemic defect of the pituitary. Their kinship, on the other hand, strongly suggests a hereditary etiology of PPE in these cases. The normal appearance of the sellar region, and the absence of PPE in both parents, makes dominant heredity of PPE unlikely.
Conclusion
To our knowledge, the presented cases of PPE are the first cases of PPE in siblings. Although no mutations were found in a genetic analysis, a hereditary etiology of PPE can be assumed. Our cases further support the theory that a congenital defect can be responsible for CIGHD. Therefore, in our opinion, MR evaluation for hypothalamic-hypophyseal abnormalities in family members of a child with pituitary dwarfism is recommended.
Footnotes
↵1 Address reprint requests to David Maintz, MD, Institut für Klinische Radiologie, Westfälische Wilhelms-Universität Münster, Albert-Schweitzer-Str. 33, D-48129 Münster.
References
- Received October 4, 1999.
- Accepted after revision January 25, 2000.
- Copyright © American Society of Neuroradiology
In this issue




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.