
Abstract
Summary: Citrullinemia is a rare autosomal recessive inborn error of the urea cycle due to a deficiency in argininosuccinic acid synthetase. We present two cases of the infantile form of citrullinemia in which CT revealed bilateral and symmetric corticosubcortical hypoattenuating areas, ulegyric changes, and atrophy in the frontal lobes, as well as atrophy in the gyrus cinguli, insulae, and temporal lobes.
Initially described by Mc Murray et al in 1962 (1), citrullinemia is a rare autosomal recessive inborn error of the urea metabolism due to a deficiency in argininosuccinic acid synthetase (AAS) (2). Three clinical presentations have been characterized as the neonatal, infantile (or subacute), and adult (or late-onset) forms (3). An alternate classification based on the enzymatic anomaly has been proposed; this system divides citrullinemia into type I (abnormal AAS), type II (decreased level of AAS), and type III (undetectable AAS level). The enzymatic anomaly can involve all the tissues that normally produce AAS (types I and III), or it can be limited to the liver (type II) (4).
We report the appearance of citrullinemia on CT scans, as documented in two patients with the infantile form of the disease, and we review the biochemical mechanism underlying this metabolic disorder.
Case Reports
Case 1
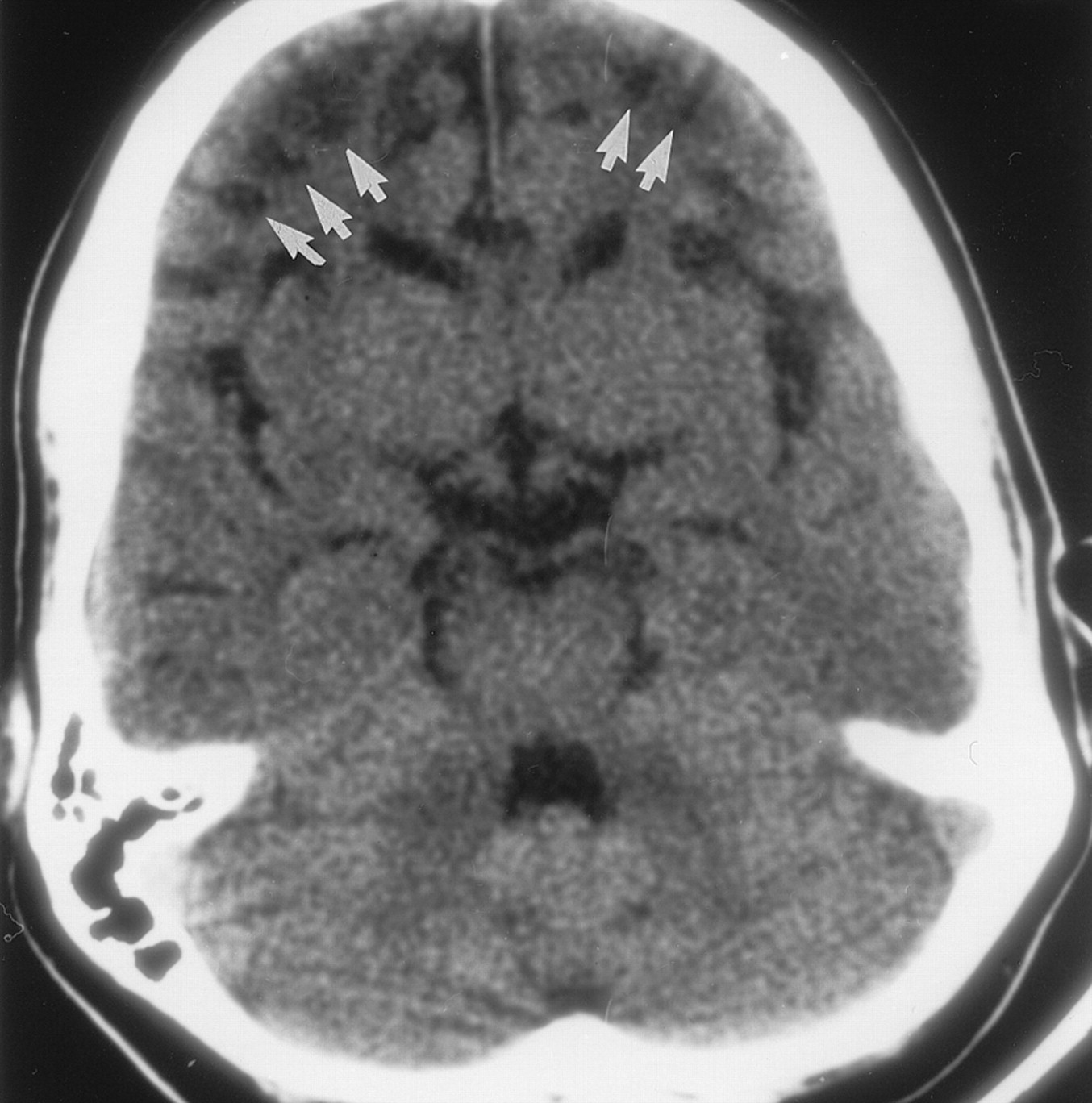
The first patient was an 8-year-old boy examined for citrullinemia that was diagnosed on the basis of AAS enzyme activity assay findings in cultured fibroblasts. His history included recurrent hospital admissions for attacks of lethargy alternating with hyperactivity; the first of these occurred when the patient was aged 4 months. He was referred to our institution for an episode of acute ataxia and vomiting. Biochemical workup revealed blood hyperosmolarity, hyperammonemia, and elevated citrulline levels, as well as an elevated orotic acid level in the urine. Aggressive management based on dialysis and rehydration, sodium benzoate administration, and dietary protein restriction was introduced. Brain CT revealed severe ulegyric changes, as well as bilateral and symmetric corticosubcortical hypoattenuating areas, with apparent sparing of the sulci apices in both frontal lobes. Brain volume loss was seen in the bilateral frontal lobes, gyri cinguli, and insulae (Fig 1). Findings in the occipital lobes, basal ganglia, thalami, and cerebellum were unremarkable.

Nonenhanced axial CT scan obtained in patient 1 demonstrates severe symmetric and bilateral ulegyric changes as well as multiple corticosubcortical hypoattenuating areas (arrows), with apparent sparing of the sulci apices in both frontal lobes. Note that the bilateral frontal lobes, gyri cinguli, and insulae are atrophic.
Case 2
The second patient was an 11-year-old boy who was initially examined when he was aged 6 months for an episode of altered mental status, vomiting, and lethargy; this was followed by multiple hospital admissions for similar attacks. The diagnosis of citrullinemia was confirmed with findings from biochemical workup that included an AAS enzyme activity assay in cultured fibroblasts. The present episode of altered mental status and lethargy was treated with dialysis and rehydration, the administration of sodium benzoate, and dietary protein restriction. Brain CT scans showed ulegyric changes and symmetric corticosubcortical hypoattenuating areas, with apparent sparing of the sulcal apices in both frontal lobes (Fig 2A). Bilateral hypoattenuating areas were seen in the claustrum. Bilateral atrophy of the cingulate gyri, insular regions, and frontal and temporal lobes was noted (Fig 2B and C). Findings in the occipital lobes, basal ganglia, thalami, and cerebellum were unremarkable.

Images obtained in patient 2.
A, Contrast-enhanced axial CT scan shows bilateral and symmetric hypoattenuating areas located in the depth of the sulci in both frontal lobes (arrows), with sparing of the cortical apices.
B, Contrast-enhanced axial CT scan shows prominent cingulate gyri atrophy and ulegyric changes in the frontal lobes.
C, Nonenhanced axial CT scans shows bilateral hypoattenuating areas in the claustrum (arrows) and atrophy of the frontal and temporal lobes.
Discussion
Citrullinemia represents the fourth most common anomaly of the urea metabolic pathway. However, its prevalence remains unspecified in the literature, which only suggests that male and female cases seem to occur equally (3). Citrullinemia is due to a deficiency in AAS, which McMurray and coauthors (1) initially described in 1962. At least three phenotypes are clinically recognized: the neonatal, infantile (or subacute), and adult (or late-onset) forms. The last form seems to be almost exclusively limited to Japanese patients. Saheki et al (4) divided the enzymatic deficiencies in patients with citrullinemia into three patterns that are characterized by present but abnormally functioning AAS (type I), normal AAS present at in abnormally low concentrations (type II), or extremely low or undetectable concentrations of AAS (type III). The enzyme deficiency in type I and type III is found in all the tissues and/or cells that normally express AAS, whereas type II involves decreased levels of AAS only in the liver; the concentration in other tissues, such as the kidney or brain, are normal (4). Type I and type III enzymatic anomalies are associated with the neonatal and infantile forms of citrullinemia, respectively, whereas type II deficiency is linked with the late-onset (or adult type) form of the disease.
Clinically, the severe neonatal form is characterized by irritability, lethargy, poor feeding, grunting, and tachypnea that occurs within the first few days of life; these subsequently progress to rigidity and sometimes opisthotonos, apnea, convulsions, coma, and death. Most patients with this form die during the neonatal period. The subacute (or infantile) form of citrullinemia causes recurrent episodes of lethargy, acute ataxia, hyperactivity, vomiting, and dehydration, which potentially lead to coma (3, 5). The late-onset form is characterized by episodes of altered consciousness, restlessness, and abnormal behavior during adulthood (6). The diagnosis of citrullinemia is based on findings from the biochemical workup, particularly AAS activity. Abnormal laboratory findings include hyperammonemia, hyperosmolarity, respiratory alkalosis, high citrulline levels in the blood, and high orotic acid levels in the urine. Low arginine blood levels and elevated glutamine, alanine, and lysine blood levels are detected during acute episodes. The definitive diagnosis of citrullinemia relies on the determination of AAS activity in liver samples and cultured skin fibroblasts. A prenatal diagnosis can be obtained by measuring the AAS activity in cultured chorionic tissue and amniocytes (3). The long-term treatment of citrullinemia consists of protein intake restriction and sodium benzoate and sodium phenylacetate administration. Peritoneal dialysis or hemodialysis is used during acute episodes. Liver transplantation has recently been proposed as another therapeutic option for citrullinemia (7). The mortality rate of citrullinemia, if left untreated, is 100%.
Publications dealing with the imaging appearance of citrullinemia are sparse, probably because the diagnosis of citrullinemia is typically based on biochemical and clinical findings. Cerebral edema has been occasionally depicted on CT scans in patients with citrullinemia, especially the neonatal form (5, 8). Japanese authors have reported the MR imaging findings in two cases of citrullinemia: One report described a small hyperintense lesion in the right cingulate gyrus on T2-weighted images (9), and the other showed hypointensities on T1-weighted images and hyperintensities on T2-weighted images in the central pontine area and both middle cerebellar peduncles (6). To our knowledge, the CT appearance of citrullinemia after multiple acute episodes or during the chronic phase of the disease has not been reported. Our first patient had severe volume loss and symmetric bilateral hypoattenuating areas in both frontal lobes. Minimal volume loss associated with ventricular enlargement was also observed in the bilateral insulae and temporal lobes. Ulegyria, a cerebral cortex anomaly due to scar tissue formation that usually occurs in early development, was present in the bilateral frontal regions. Our second patient had similar ulegyric changes, well-defined corticosubcortical hypoattenuating areas, and moderate volume loss in both frontal lobes. Severe atrophy was noted in the bilateral temporal lobes and insular regions. Additionally, hypoattenuating areas were seen in both claustra.
Our imaging findings correlate with the neuropathologic findings in citrullinemia described by Martin et al in 1982 (10). These authors reported the presence of frontoparietal ulegyria with relative sparing of the mediotemporal gyri and occipital lobes; corticosubcortical areas of necrosis with the formation of microcavities; ventricular dilatation; and preservation of the basal ganglia, brain stem, and cerebellum. Histologic studies showed multiple cortical necroses ranging from pseudolaminar neuronal losses to areas of complete depopulation, with hyperplasia of endothelial cells, a large amount of macrophages filled with neutral fat, and fibrillary astrocytes. Although incomplete parenchymatous necrosis was often present in the crown of the gyri, more severe necrotic changes were always found in the depth of the sulci and in the adjacent walls of the convolutions (10). These findings are consistent with our CT observations of bilateral, symmetric, corticosubcortical hypoattenuating areas with apparent sparing of the sulci apices (Fig 2A). Findings from necropsy in a deceased patient with the adult form of the disease (type II) confirmed the preferential involvement of the cortex located in the depth of the sulci in the frontal, insular, occipital and left temporal regions. In this case, the cerebral white matter was mostly preserved, although edema was present (11).
Both the ulegyric and ischemic areas had selective neuronal loss, with relative sparing of the watershed areas and Purkinje cells. This finding suggests that the changes observed in both types of lesions are not caused by a global ischemic mechanism, but rather, by local hypoperfusion that is probably secondary to arterial involvement at the level of the subarachnoid space (11). Okken et al (12) have shown in animal models that the administration of citrulline inhibits the aerobic metabolism of glucose. This observation, as well as that in a reported necropsy case involving the neonatal form with typical pathologic changes in citrullinemia but without associated hyperammonemia, suggests that citrulline has a role as the causative agent of the disease. The severity of the neuropathologic changes is also correlated with the concentration of citrulline in the blood and CSF (1).
Conclusion
We report the CT findings in two patients with the infantile form of citrullinemia. In particular, scans showed severe ulegyric changes and atrophy of the frontal lobes, as well as bilateral and symmetric corticosubcortical frontal hypoattenuating areas. These findings are consistent with reported neuropathologic findings in cases of citrullinemia. CT is easily performed in the affected patient population (small children), and the reported characteristics may prove useful in the diagnosis and follow-up of patients with citrullinemia.
- Received November 17, 2000.
- Accepted after revision January 9, 2001.
- Copyright © American Society of Neuroradiology





{kind=link}
{kind=link}