
Abstract
SUMMARY: Xanthoma disseminatum is a rare normolipemic histiocytic disorder of non-Langerhans cell origin. It is a chronic systemic disease with a benign course, characterized by disseminated, yellow-orange–colored papules on the face, flexures, and mucosal membranes. We report 3 patients with xanthoma disseminatum, who presented primarily with central nervous system disease and a multitude of imaging findings throughout the craniospinal axis.
Xanthoma disseminatum affects different age populations, and cases have been reported between the ages of 5 months and 70 years. The disease is more prevalent in males with a male-to-female ratio of 2:1. No typical inheritance pattern or geographic distribution is recognized.1 Three clinical variants are described on the basis of disease progression and prognosis: a common persistent form, a less common progressive form with systemic involvement, and a rare form of self-limited disease. Central nervous system (CNS) involvement is rare and usually occurs in the systemic variety.2,3 Xanthoma disseminatum is characterized by a normal lipid profile.1,4 Diabetes insipidus is a common manifestation of xanthoma disseminatum, related to extensive meningeal involvement in the region of the sella and hypophysis.5–9
Case 1
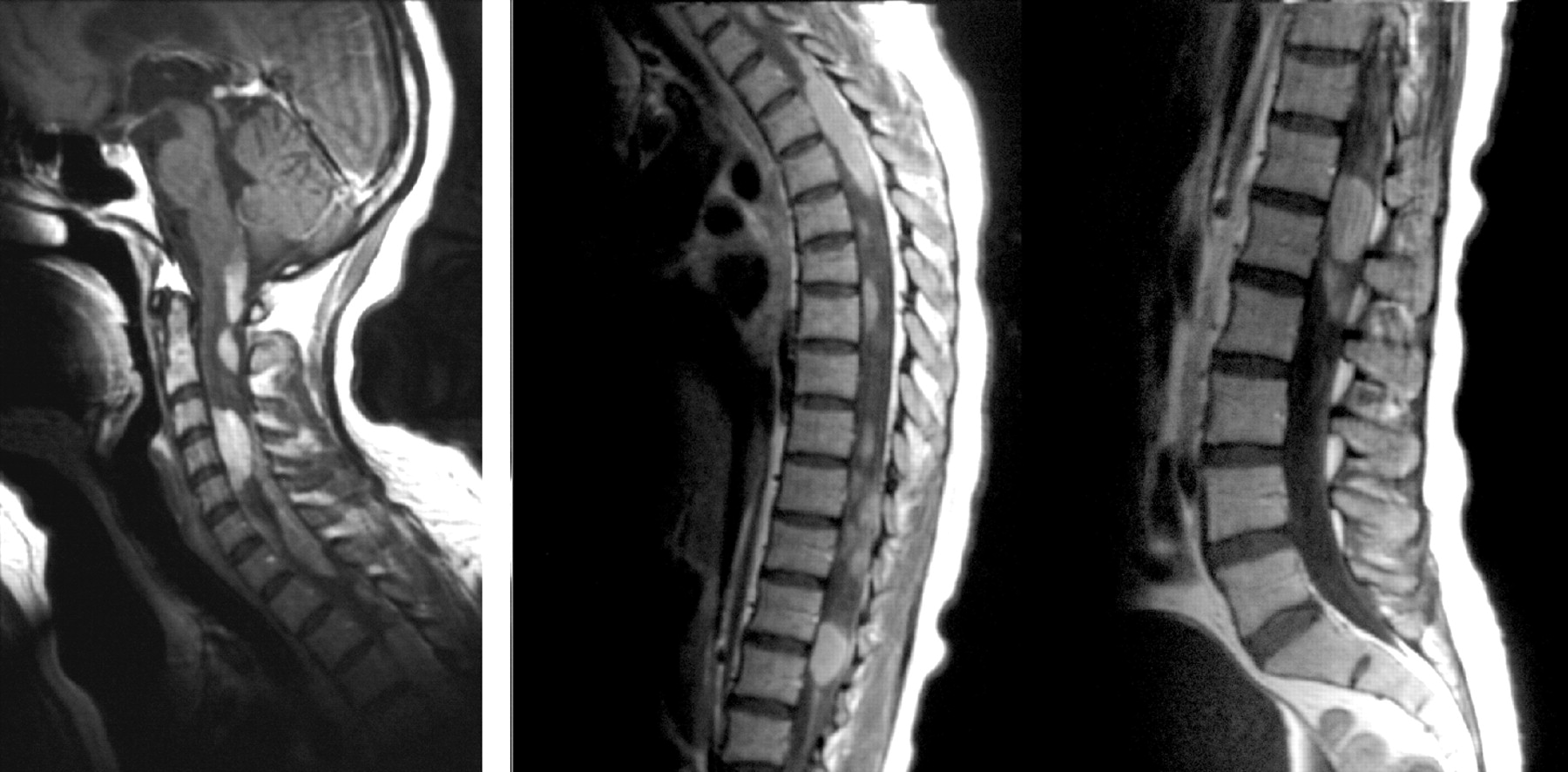
A 23-year-old white woman presented with partial complex seizure in the left upper extremity. Initial work-up disclosed a normal biochemical profile. A brain MR imaging examination on admission showed a 3-cm round heterogeneously enhancing mass in the right thalamus with a small amount of vasogenic edema and mild local mass effect (Fig 1). The patient underwent complete surgical excision of the tumor. Histopathologic findings suggested the histiocytic nature of the tumor, and additional histochemical analysis confirmed a diagnosis of xanthoma disseminatum. Three months later a follow-up MR imaging examination showed local recurrence of the tumor. A second surgery was performed, and the mass was partially removed. Radiation therapy was administered. Twelve months later, the patient presented with progressive weakness in the extremities and inability to stand upright. MR imaging examination of the brain showed further progression of the tumor with new lesions seen in the brain stem and proximal spinal cord. MR imaging of the entire spine showed extensive patchy T2 signal intensity abnormalities throughout the brain stem and spinal cord. Postgadolinium images showed intensely enhancing variable-sized intra- and extra-axial lesions (Fig 2). The patient died 2 years after initial presentation.

Case 1: A, Axial T1-weighted image shows a large heterogeneous mass lesion in the right thalamus with mild local mass effect.
B, Axial T2-weighted image shows a poorly defined heterogeneous mass with a small amount of vasogenic edema.
C, Axial T1-weighted image postgadolinium shows intense and heterogeneous enhancement of the mass lesion.

Case 1. Sagittal T1-weighted image of the whole spine shows multiple intra- and extra-axial lesions with intense enhancement.
Case 2
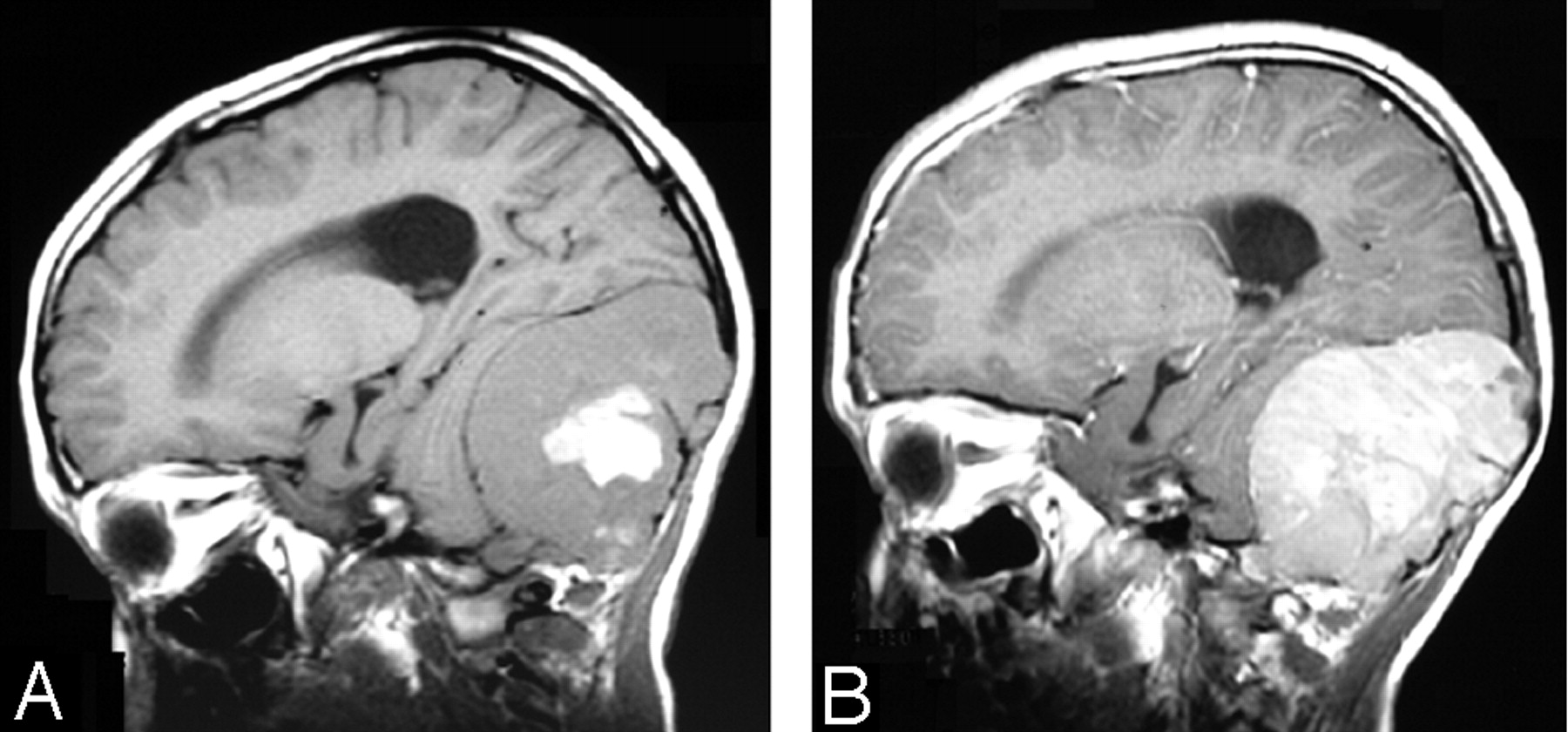
A 13-year-old black boy presented with chronic headache and dizziness. The patient was otherwise healthy. Results of complete blood counts and biochemical profiles were normal. Initial brain MR imaging examination showed a large heterogeneous extra-axial mass lesion with intense enhancement. There was considerable mass effect on the cerebellum and fourth ventricle (Fig 3). The tumor was completely excised. Four years later, the patient continues to do well with mild encephalomalacic changes in the cerebellum and no evidence of tumor recurrence.

Case 2. A, Sagittal T1-weighted image shows a large heterogeneous extra-axial mass in the posterior fossa with central hemorrhage.
B, Sagittal T1-weighted image shows intense enhancement of the tumor. Notice the relationship of the tumor to the dura and the lack of a dural tail sign.
Case 3
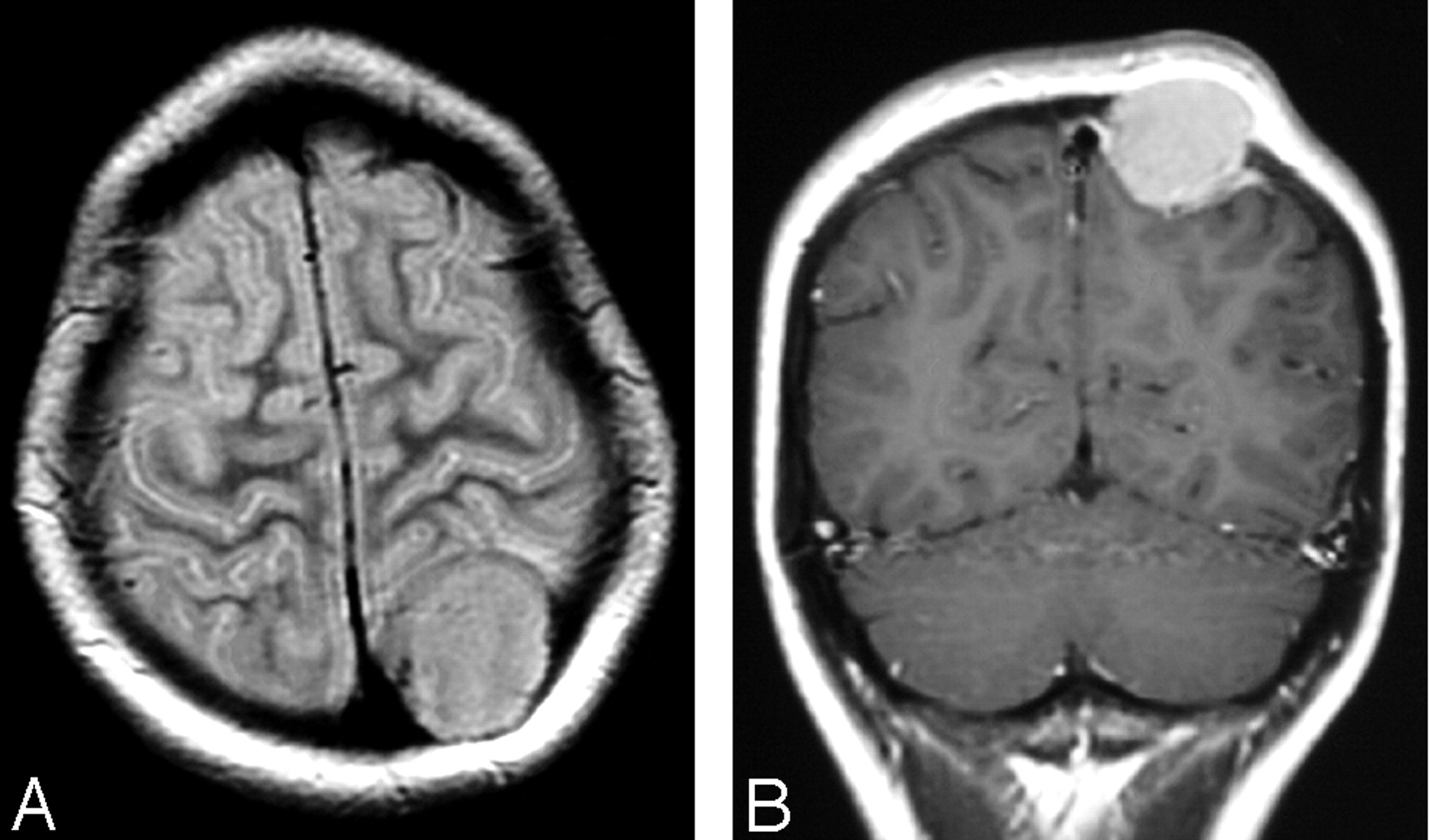
A 14-year-old white girl presented with a painless bump on her head. There were no other complaints and no neurologic deficits. Results of complete blood counts and biochemical profiles were normal. A brain CT scan was initially performed and showed a well-defined osteolytic lesion in the left parietal bone. A brain MR imaging was subsequently performed and confirmed the presence of an intensely enhancing calvarial tumor that had violated the inner and outer tables (Fig 4). A whole-body radionuclide bone scan showed radiotracer uptake around the tumor in the left parietal bone without additional abnormalities. Two years later the patient remains free of disease.

Case 3. A, Axial T2-weighted image shows minimal vasogenic edema in the outer aspect of the adjacent parietal lobe.
B, Coronal T1-weighted image postgadolinium shows intense enhancement and a dural tail sign.
Discussion
CNS involvement in histiocytic disorders is unusual and includes forms of Langerhans cell histiocytosis, plasma cell granuloma, Rosai-Dorfman disease, Erdheim-Chester disease, malignant fibrous histiocytoma, histiocytic lymphoma, intracranial xanthoma associated with hyperlipidemia, and xanthoma disseminatum. The distinction between different histiocytic disorders became possible with modern immunohistochemical techniques and electron microscopy.1,4-6 Xanthoma disseminatum was described as a distinct histiocytic disorder by Albrecht von Graefe in 1867. The disease is also known as Montgomery syndrome and xanthoma multiplex.4,6
Xanthoma disseminatum is characterized by large pleomorphic multinucleated histiocytes with foamy cytoplasm immunoreactive for CD68. The numerous histiocytes are intermixed with variable numbers of mature T-lymphocytes, immunoreactive for leukocyte common antigens and CD3. Atypia, mitoses, and necrosis are typically not present. No significant inflammatory component is seen (Fig 5). Histiocytic cells of xanthoma disseminatum are not immunoreactive for glial fibrillary acidic protein, which characterizes pleomorphic xanthoastrocytoma. Unlike other more common forms of histiocytosis, the histiocytic cells of xanthoma disseminatum are not immunoreactive for S-100. Birbeck granules, characteristic of Langerhans cell histiocytosis, are not seen on electron microscopy.1,4-6

Photomicrographs of a specimen from case 1 (Courtesy of William Kupsky, MD, Wayne State University, Detroit, Mich).
A, Hematoxylin and eosin stain (×400) shows numerous irregular large cells with abundant foamy cytoplasm and vesicular nuclei.
B, Xanthoma disseminatum cells stain (×400) strongly positive for the CD68 marker.
MR imaging of the brain shows a well-defined heterogeneous and intensely enhancing intra-axial mass lesion. There is typically some local mass effect without significant vasogenic edema (case 1). A recently published case report described a patient with xanthoma disseminatum who presented with imaging features mimicking cerebral glioma.10 Extra-axial lesions are characterized by a well-circumscribed outline with intense enhancement and lack of the “dural tail” sign (case 2). Bony disease presented with sharply defined osteolytic and intensely enhancing geographic lesions, and bone scintigraphy showed mild radiotracer uptake (case 3). Expansile cystic lesions in the small bones of the hands have been reported.11 MR imaging of the spine showed patchy high T2-signal-intensity abnormality in the brain stem and throughout the spinal cord without hemorrhage or syringomyelia. Postgadolinium images showed intense enhancement of multiple intra- and extra-medullary lesions (case 1).
Demyelination may occur in some histiocytic disorders. Lesions resembling multiple sclerosis have been described in Erdheim-Chester disease.12,13 In our experience, demyelinating lesions were not seen in patients with xanthoma disseminatum. In the medical literature, the term “xanthoma disseminatum” has been used loosely, and the disease has been confused with other entities. Some case reports of what has been referred to as xanthoma disseminatum of the CNS could fit into other histiocytic disorders, such as Rosai-Dorfman disease. Only a few case reports labeled with this diagnosis were based on modern histochemical techniques. To our knowledge, the disease has never been described in the spinal cord, and our first case is unique in this regard.
Local control of the disease can be achieved with complete surgical excision of the offending tumor. Low-dose radiation therapy (1000 cGy in doses of 200 cGy daily) is known to be beneficial in inflammatory histiocytic disorders, such as Rosai-Dorfman disease. However, it appears that xanthoma disseminatum is radioresistant.14 In some patients with systemic disease, cyclophosphamide was used to control severe symptoms such as stridor, caused by tracheal involvement, and loss of vision, caused by severe periorbital disease, but no recovery of pituitary function was reported after chemotherapy.9,15 We encountered a patient with xanthoma disseminatum who continued to experience local recurrence that progressed to involve the entire neural axis, despite multiple attempts at complete surgical excision and radiation therapy (case 1).
References
- Received March 22, 2005.
- Accepted after revision June 3, 2005.
- Copyright © American Society of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}