
Abstract
SUMMARY: SGPL1 encodes sphingosine-1-phosphate lyase, the final enzyme of sphingolipid metabolism. In 2017, a condition featuring steroid-resistant nephrotic syndrome and/or adrenal insufficiency associated with pathogenic SGPL1 variants was reported. In addition to the main features of the disease, patients often exhibit a range of neurologic deficits. In a handful of cases, brain imaging results were described. However, high-quality imaging results and a systematic analysis of brain MR imaging findings associated with the condition are lacking. In this study, MR images from 4 new patients and additional published case reports were reviewed by a pediatric neuroradiologist. Analysis reveals recurring patterns of features in affected patients, including isolated callosal dysgenesis and prominent involvement of the globus pallidus, thalamus, and dentate nucleus, with progressive atrophy and worsening of brain lesions. MR imaging findings of abnormal deep gray nuclei, microcephaly, or callosal dysgenesis in an infant or young child exhibiting other typical clinical features of sphingosine-1-phosphate lyase insufficiency syndrome should trigger prompt genetic testing for SGPL1 mutations.
ABBREVIATIONS:
- PKAN
- pantothenate kinase-associated neurodegeneration
- S1P
- sphingosine-1-phosphate
- SPLIS
- sphingosine-1-phosphate lyase insufficiency syndrome
Sphingosine-1-phosphate lyase insufficiency syndrome (SPLIS), also referred to as nephrotic syndrome type 14, is a recently discovered severe inborn error of metabolism caused by biallelic pathogenic variants of SGPL1, the gene encoding sphingosine-1-phosphate lyase.1 Sphingosine-1-phosphate lyase is an intracellular enzyme responsible for catalyzing the final step in sphingolipid degradation, in which the universal sphingolipid breakdown product sphingosine-1-phosphate (S1P) is irreversibly degraded (Fig 1).2 S1P is a bioactive lipid that mediates signals through a family of G protein-coupled receptors that regulate lymphocyte trafficking, vascular integrity, and other physiologic and developmental processes.3,4

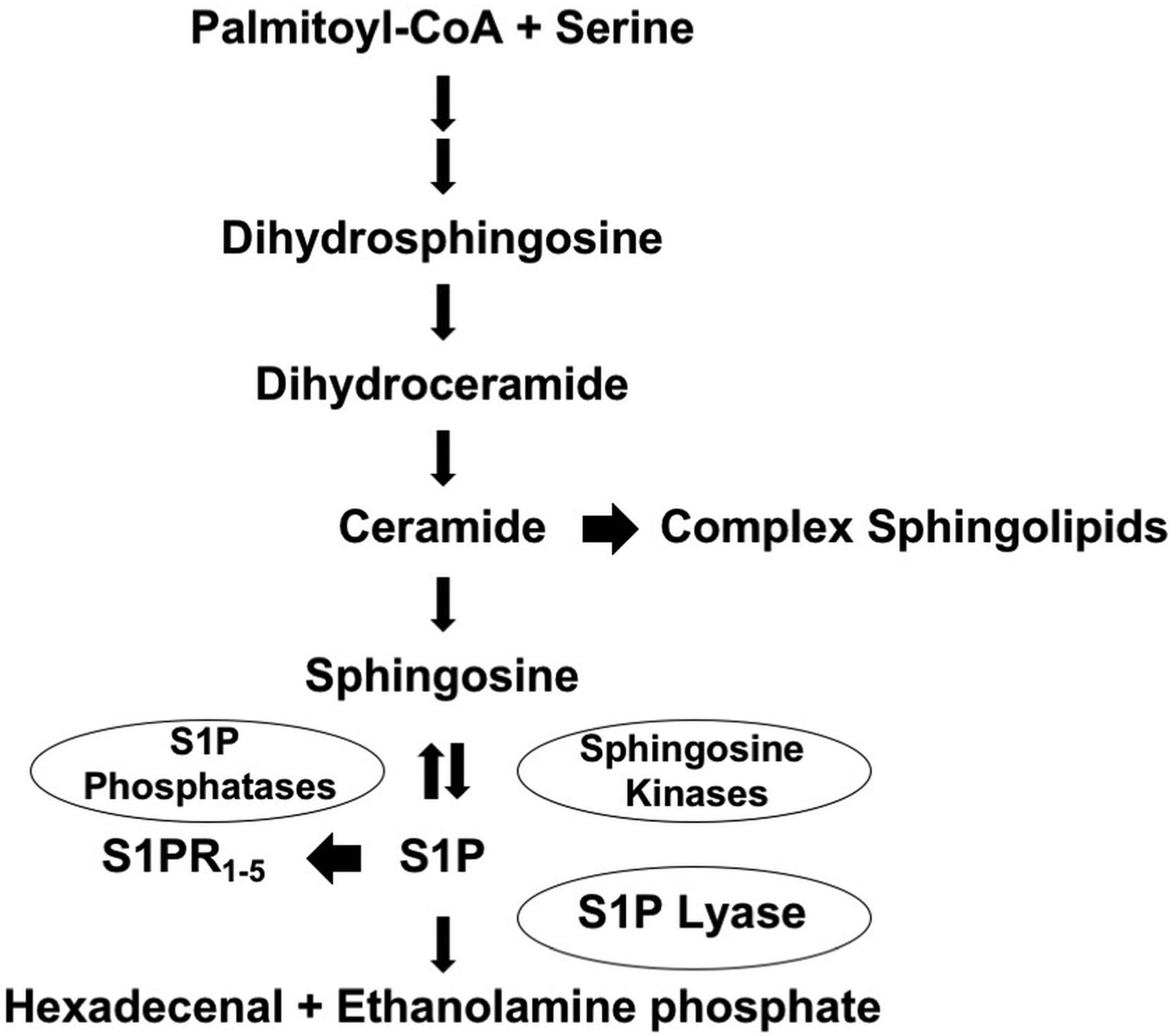
Sphingolipid metabolic pathway. Sphingolipid biosynthesis leads to the formation of ceramide, the main building block of complex sphingolipids. Ceramide is degraded to sphingosine. Sphingosine is phosphorylated by sphingosine kinases, leading to formation of S1P, which has intracellular activities and also serves as a ligand for S1P receptors (S1PR1–5). S1P can be dephosphorylated by specific and nonspecific lipid phosphatases, regenerating sphingosine. Alternatively, S1P can be irreversibly degraded by sphingosine-1-phosphate lyase (S1P lyase), which controls the sole exit point of sphingolipid metabolism. S1P catabolism by sphingosine-1-phosphate lyase results in formation of hexadecenal and ethanolamine phosphate. CoA indicates coenzyme A.
The first descriptions of a human syndrome of sphingosine-1-phosphate lyase insufficiency associated with inactivating mutations in SGPL1 and sphingolipid accumulation were reported in 2017, and during the subsequent 2 years, approximately 45 patients have been identified in total worldwide (including our unpublished cohort of 7 new patients).5⇓⇓⇓⇓⇓-11 A wide spectrum of severity has been observed, ranging from nonimmune hydrops fetalis to survival into at least the third decade. However, most patients with SPLIS exhibit severe manifestations of ≥1 of the main clinical features including steroid-resistant nephrotic syndrome, primary adrenal insufficiency, lymphopenia, ichthyosis, hypothyroidism, micropenis, and neurologic defects.
Neurologic abnormalities were reported in >50% of the cases. The incidence of neurologic complications may be higher than reported because neurologic status could not be determined in cases resulting in fetal loss or in patients requiring basic life support. Neurologic manifestations may include macrocephaly, microcephaly, hypotonia, cranial nerve palsies, sensorineural hearing loss, seizures, Charcot-Marie-Tooth-type peripheral neuropathy, ataxia, developmental delay and/or regression, developmental brain anomalies, and severe neurologic deterioration resulting in death.
Brain imaging results have been reported for some patients with SPLIS (On-line Table). Lesions including cortical atrophy; contrast enhancement of the midbrain, pons, and/or cerebellum; and developmental anomalies were described. We have performed a detailed study of 4 new patients with genetically confirmed SPLIS including 1 infant in whom the progressive nature of the disease was evident on serial MR imaging. Interpreted in the context of other reported cases of SPLIS, our findings reveal several patterns of MR imaging changes that may aid in the recognition of this novel and atypical (nonlysosomal) sphingolipidosis.
MATERIALS AND METHODS
The main focus of this study is the MR images of the brain from 4 new unrelated patients with biallelic SGPL1 pathogenic variants identified by whole-exome sequencing and referred to us from the King Saud University College of Medicine in Saudi Arabia, the Cincinnati Children’s Hospital Medical Center in the United States, and the Harran University Department of Medicine in Turkey. Individuals had testing that was interpreted by a College of American Pathologists/Clinical Laboratory Improvement Amendments–certified molecular testing lab. Pathogenicity calling followed the American College of Medical Genetics guidelines. MR imaging studies were systematically reviewed by a qualified pediatric neuroradiologist. Images from a fifth patient who had been previously reported were obtained for review.5 CT imaging, when available, did not reveal abnormalities even when MR imaging findings for the same patient at the same time point were abnormal. We have not included these negative CT results in our report. We retrospectively reviewed MR images from published case reports by Lovric et al,5 Prasad et al,6 Bamborschke et al,10 and Settas et al.11 The study was conducted in accordance with human subject protocols and consent forms that were reviewed and approved by the institutional review boards of UCSF Benioff Children’s Hospital Oakland, Cincinnati Children’s Hospital Medical Center, King Saud University College of Medicine, and the Harran University Department of Medicine.
Patients
The first patient, from Saudi Arabia, was homozygous for c.1079G>T; p. Gly360Val SGPL1 pathogenic variant as determined by whole-exome sequence analysis at 3 years of age. She developed a squint at 1 year of age requiring surgical correction, steroid-resistant nephrotic syndrome at 3 years of age, and left-radial neuritis several months later. At 4 years of age, she lost the ability to walk or sit with support. Despite treatment for presumed autoimmune encephalitis with intravenous immunoglobulin, high-dose prednisolone, baclofen, biotin, trihexphenydyl, clonazepam, and, later, pyridoxine, she continued to deteriorate from chronic progressive encephalopathy. Her course was further complicated by suppression of the thyroid gland due to biotin, which resolved without medication. She also exhibited lymphopenia and was being investigated for adrenal insufficiency. However, testing for the latter could not be completed because she was on high corticosteroid doses. She died of presumed sepsis at 4.5 years of age. MR imaging was performed at 4 years of age.
The second patient, from Saudi Arabia, was homozygous for c.665G>A; p.Arg222Gln SGPL1 pathogenic variant as determined by whole-exome sequence analysis at 9 years of age. The patient presented at 6 years of age with convulsions, electrolyte disturbance, and hyperpigmentation and was diagnosed with ichthyosis and adrenal insufficiency, which was treated with fludrocortisone and hydrocortisone. The patient exhibited proteinuria at first presentation, followed by fluctuating renal dysfunction during the next 3 years. Renal biopsy at 9 years of age revealed focal segmental glomerulosclerosis. Dialysis was instituted due to chronic renal failure at that time. He also exhibited lymphopenia. Other than seizures at presentation, the patient had no neurologic problems until he was diagnosed with severe left carpal tunnel syndrome at 6 years of age with persistent severe weakness in the left arm. The patient remains alive and is scheduled for kidney transplantation at the time of this report. MR imaging was performed at 6 years of age.
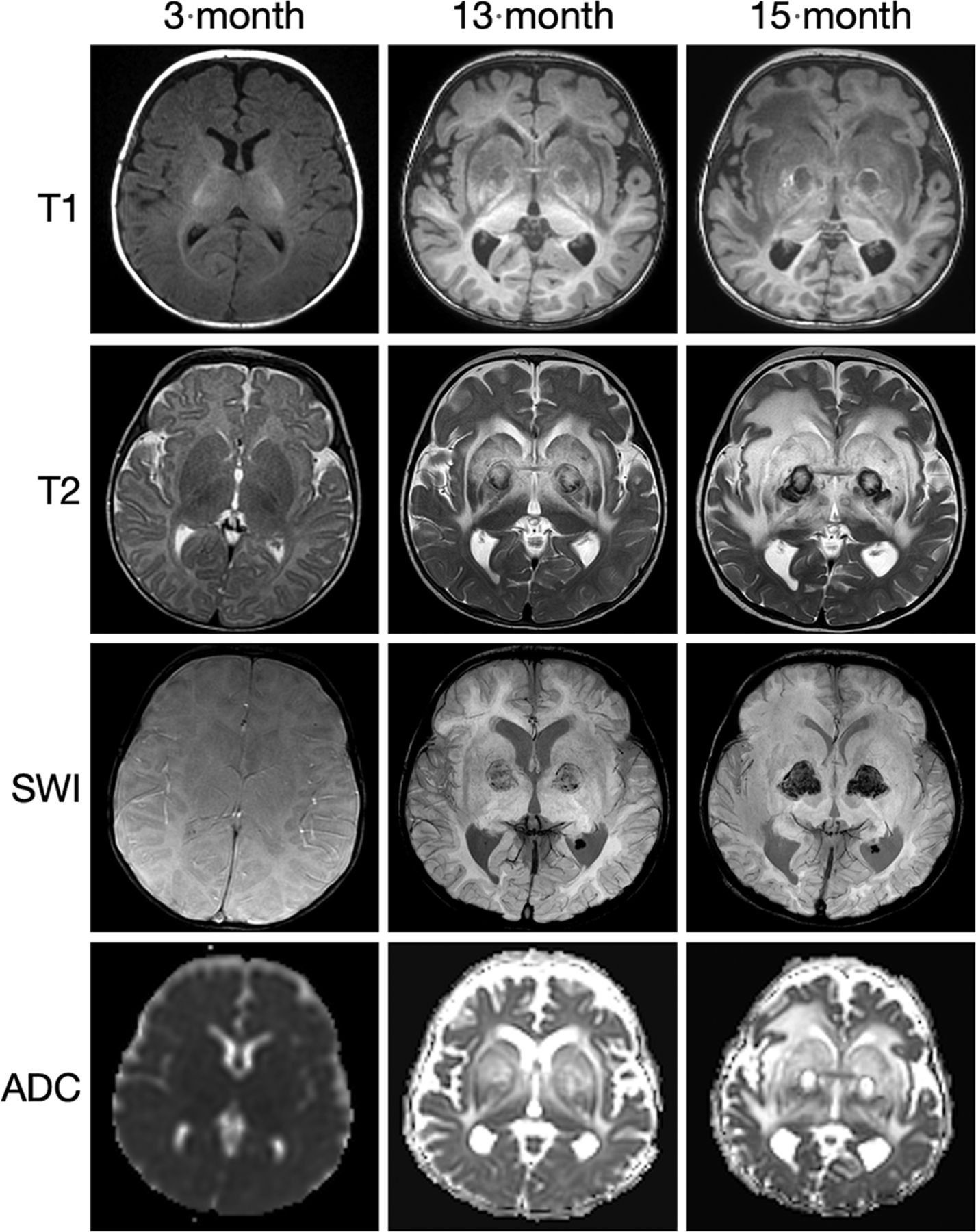
The third patient, from the United States, was compound heterozygous for c.868-T > C; p.Phe290Leu; c.993C>G; p.Tyr331* SGPL1 pathogenic variants as determined by whole-exome sequence analysis at 13 months of age. This patient had adrenal calcifications and polyhydramnios on fetal sonography imaging. Prenatal MR imaging findings of the brain were normal. His perinatal course was complicated by apnea, bilateral hearing loss, immunodeficiency including lymphopenia, and nephrotic syndrome accompanied by massive kidney enlargement. The latter required bilateral nephrectomy and peritoneal dialysis at 3 months of age. At 1 year of age, he was diagnosed with adrenal insufficiency and esotropia. At 13 months, the patient began exhibiting developmental regression. His neurologic status deteriorated steadily with intermittent seizures until his death at 17 months of age. MR imaging of the brain was performed at 25 weeks’ gestation, and postnatally at 6 weeks, 13 months, and 15 months of age.
The fourth patient, from Turkey, was homozygous for c.44A>G; p.Tyr15Cys SGPL1 pathogenic variant as determined by whole-exome sequence analysis at 2 years of age. The parents were consanguineous, and the presence of the pathogenic variant in each parent was confirmed by Sanger sequencing. The patient had a healthy infancy, but experienced a fall from the bed at 1 year of age, resulting in a hospital emergency department visit, with no traumatic injuries found. At 2 years of age, she was noted to have macrocephaly, ptosis, loss of developmental milestones, hypotonia, and incontinence. Dysmorphic features and abnormal dentition, including structural and numeric dental problems and a dentinogenesis imperfecta-like appearance, were noted. The patient remains alive at the time of this report. MR imaging of the brain was performed at 1 and 2 years of age.
Additional clinical information related to these cases is being provided separately in a therapy-related report.12
MR Imaging
MR imaging was performed on a variety of scanners using T1- and T2-weighted sequences, FLAIR, and occasionally more sophisticated methods, including diffusion-weighting, susceptibility-weighting, and postcontrast imaging. Complete studies were not available for our review. Imaging studies consisted mostly of selected key images that had been submitted with other medical data for clinical consultation. Available images from each patient were reviewed and compared.
RESULTS
Our 4 new patients were combined with 11 patients from prior publications that included patients with SPLIS who had imaging of the brain. The clinical and imaging features are summarized in the On-line Table. Five of these 15 patients had cortical atrophy or clinical findings of microcephaly. Five had edema, calcification, necrosis, or atrophy of the globus pallidus, caudate, putamen, or thalamus. Seven patients exhibited involvement of the pons or cerebellum. Gadolinium contrast, when administered, typically caused enhancement of the affected deep gray nuclei or central structures in the midbrain, medulla, and cerebellum, including the olivocerebellar circuits. The 3 patients who underwent CT scanning all reportedly had normal findings, suggesting the insensitivity of this method to detect the earliest brain abnormalities observed in patients with SPLIS.
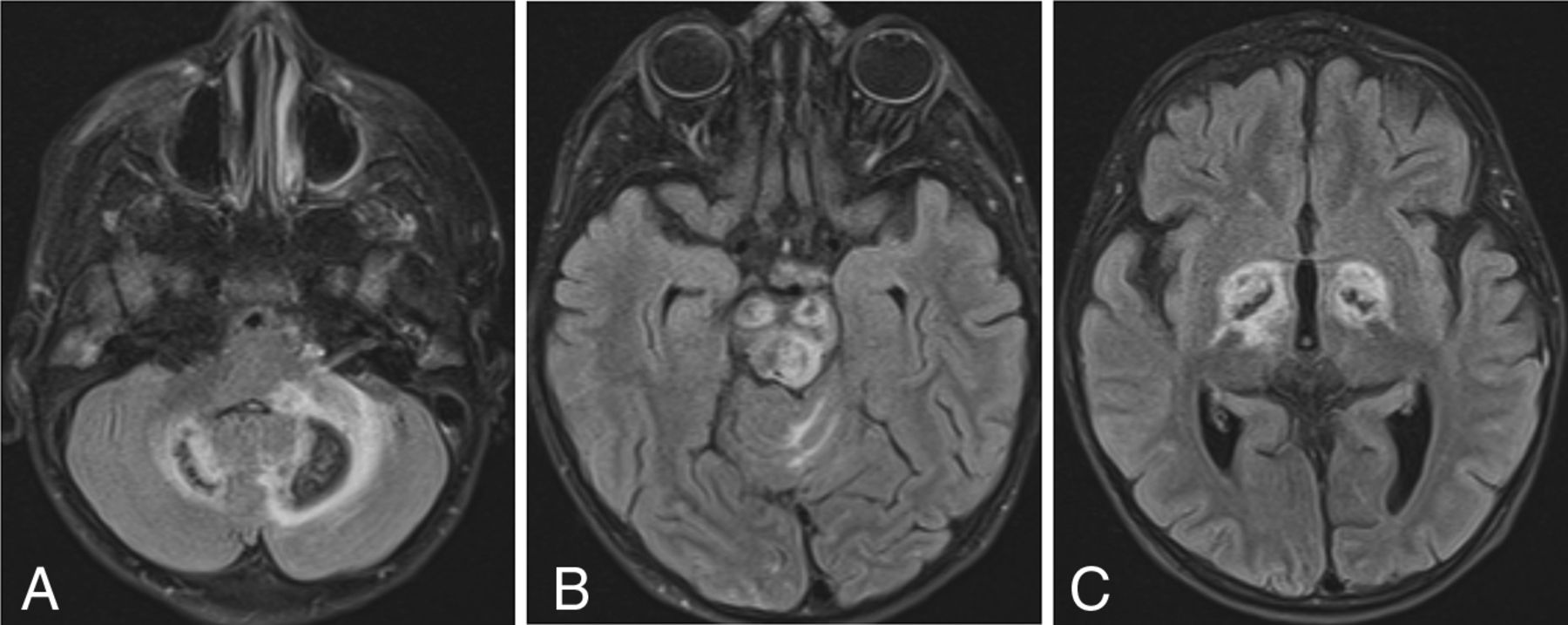
New patient 1 had 3 scans showing similar-but-progressive findings. Deep gray nuclear involvement was present, including the globus pallidus, thalamus, substantia nigra, and dentate nuclei of the cerebellum, all of which were associated with extensive surrounding FLAIR hyperintensity, presumably representing edema (Fig 2). Across time, the lesions appeared to consolidate and edema diminished, and there was a loss of central or peripheral T2 signal. Brain stem involvement was present, which could not be separated into primarily gray or white matter disease.

SPLIS with mineral deposition and edema. T2-FLAIR axial MR images of a 4-year-old child with SPLIS show involvement of the dentate nuclei (A), cerebral peduncles and tectum (B), and globi pallidi (C). There is surrounding edema. Loss of central signal is attributed to calcium on the basis of the analysis of susceptibility-weighted images in another patient. The findings predominantly involve dopaminergic neurons.
New patient 2 had a single scan at 6 years of age showing multiple small areas of cerebral and cerebellar cortical T2-FLAIR hyperintensity, some of which extended into the adjacent subcortical white matter (Fig 3). The deep gray nuclei and deep white matter structures appeared completely spared.

SPLIS with cerebral and cerebellar cortical lesions. T2-FLAIR axial images of an 8-year-old child with SPLIS show multiple focal hyperintense lesions involving cerebellar and cortical gray matter with some lesions extending into the subcortical white matter (B and C).
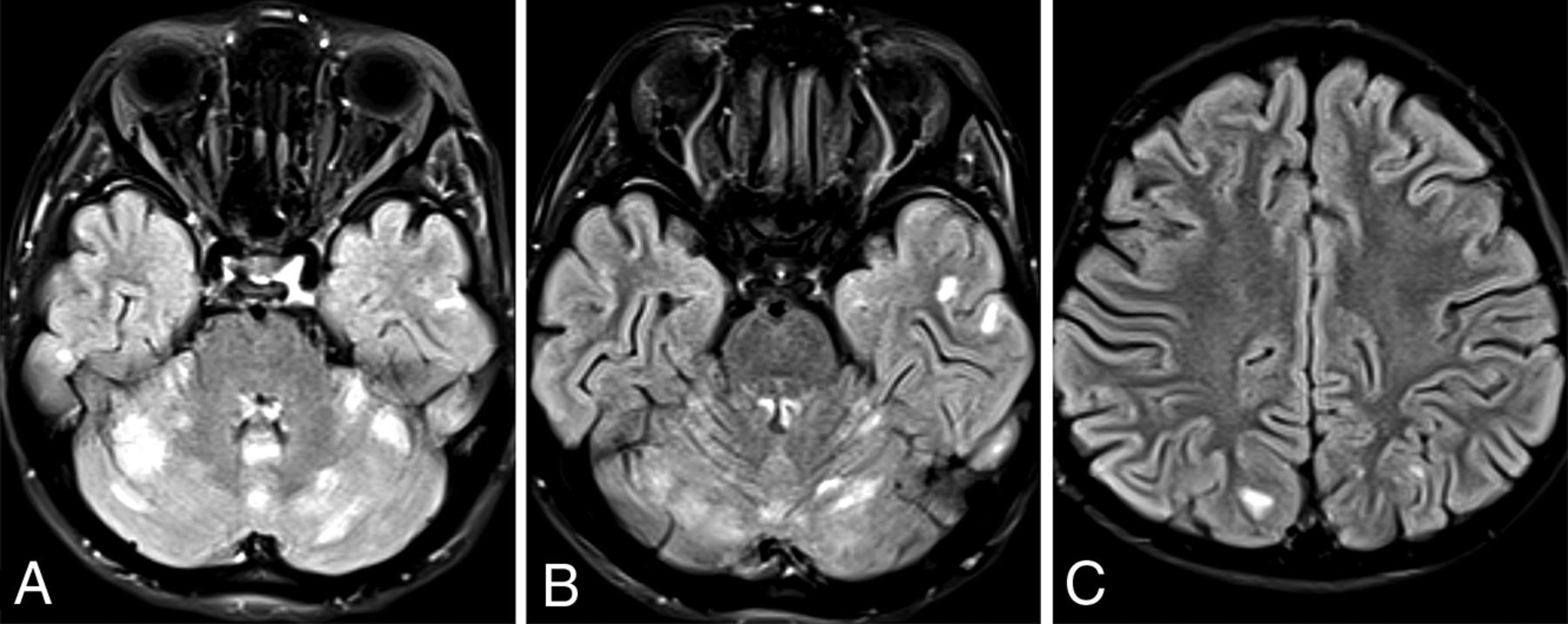
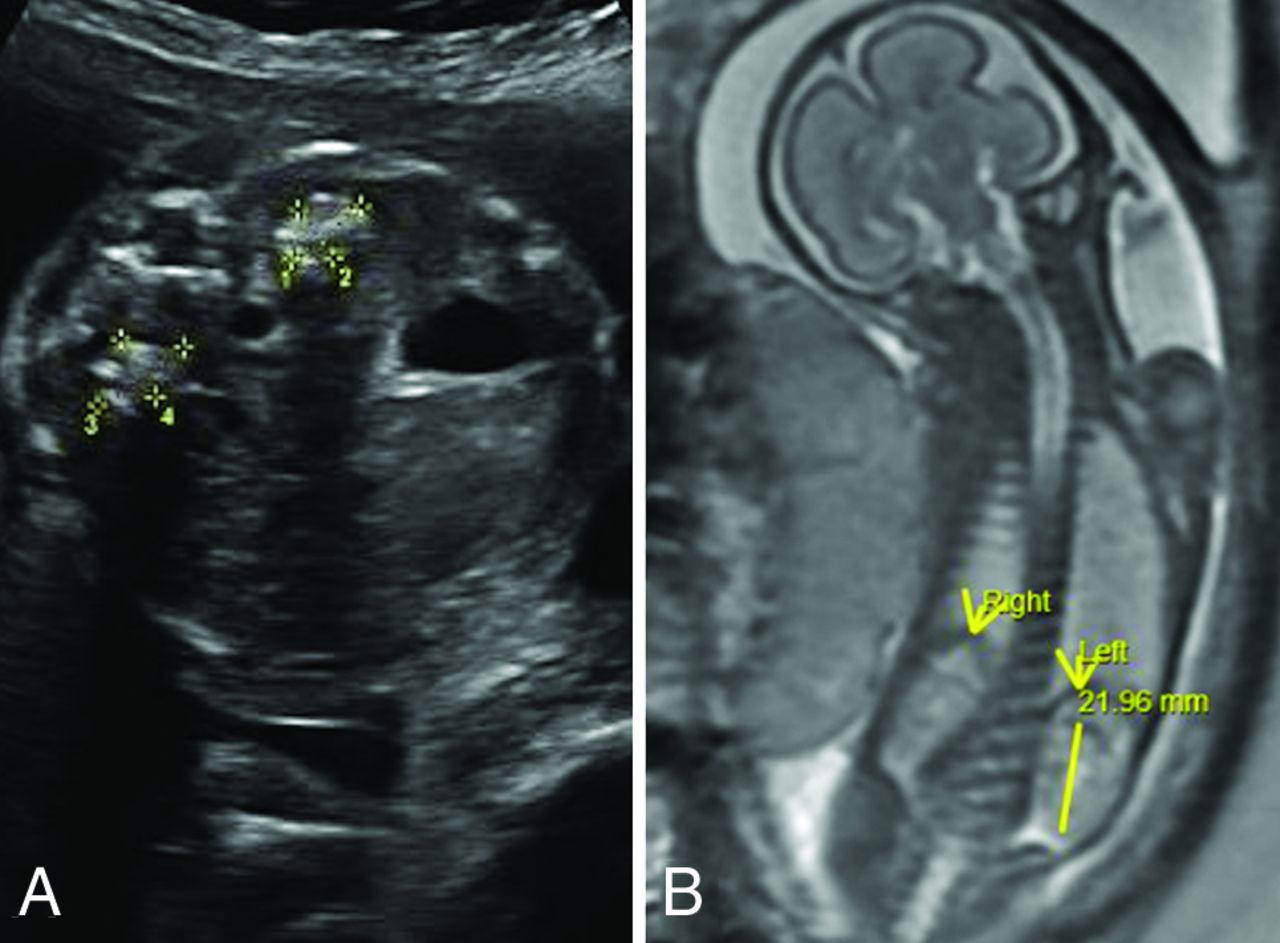
New patient 3 had fetal imaging at 25 weeks’ gestation. Sonography demonstrated enlarged echogenic adrenal glands with posterior shadowing. The concurrent fetal MR images showed prominent adrenal glands with a normal signal character suggesting calcification rather than hemorrhage (Fig 4). Postnatal MR imaging was performed at 6 weeks, 13 months, and 15 months of age (Fig 5). Initial brain imaging findings at 6 weeks were normal. At 13 and 15 months, there was extensive and progressive bilateral caudate, putaminal, and thalamic involvement, which appears to be centered on the globus pallidus. A rim of T1- and T2-shortening developed about the en-larged globi pallidi associated with the susceptibility effect and increased diffusivity. Analysis of phase images suggests the presence of calcium rather than iron.

SPLIS at 25 weeks’ gestation. Fetal sonography shows a transverse image of the abdomen (A) with echogenic enlarged adrenal glands with posterior shadowing presumably due to calcification. Fetal whole-body MR imaging (B) shows normal-appearing kidneys and adrenal glands. Mineralization of the adrenal glands is not apparent on this T2-weighted sequence.

SPLIS appearance and progression of MR imaging findings with age. Sequential MR imaging performed in this infant with SPLIS between 6 weeks and 15 months of age has initially normal findings. At 13 and 15 months of age, there is progressive involvement of the globus pallidus with extensive edema of the caudate, putamen, and thalamus. Diffusivity is increased, and there is a susceptibility effect with a ring of T1- and T2- shortening. The susceptibility-phase images indicate the presence of calcium.
New patient 4 underwent MR imaging of the brain at 1 and 2 years of age. There was callosal dysgenesis with absence of the rostrum, genu, and splenium. Otherwise, the T2-weighted images of the brain and progression of myelin maturation appeared normal at 1 and 2 years of age (Fig 6).

SPLIS with dysgenesis of the corpus callosum and hypoplasia with an absent rostrum, genu, and splenium. This 2-year-old patient with SPLIS has dysgenesis of the corpus callosum. The sagittal T1- and axial T2-weighted MR images show absence of the rostrum, genu, and splenium (A). The body of the corpus callosum is hypoplastic. Brain volume, gyral complexity, and myelin maturation are grossly normal (B).
DISCUSSION
SPLIS is a newly recognized nonlysosomal sphingolipidosis caused by recessive mutations in the gene encoding S1P lyase, an intrinsic protein of the endoplasmic reticulum outer membrane. Analysis of these patients demonstrates the variety of neuroimaging findings in SPLIS. Appearances included callosal agenesis, dysgenesis, or hypoplasia or a rapidly progressive neurodegenerative disease in other patients. The developmental malformation could range from the callosal dysgenesis alone to generalized microcephaly, or it could result in widespread gyral simplification and hypoplasia of the frontal and temporal lobes, as described by Bamborschke et al.10 Patient 4 does not exhibit typical features of SPLIS, and sphingolipid accumulation was not observed in the patient’s plasma (our unpublished findings). Thus, the clinical and imaging findings observed in this case could potentially have another etiology. Considering that another reported patient with SPLIS with biochemically confirmed pathogenic biallelic SGPL1 variants also exhibited callosal dysgenesis,5 we included this patient in our analysis. Both patients with callosal dysgenesis were Turkish, but they do not share SGPL1 pathogenic variants. This feature raises the possibility of a separate genetic defect as the cause of the developmental malformation.
Two of our patients and a total of 5 of the 15 patients reviewed in this report had progressive damage to the deep gray nuclei, including the thalamus, globus pallidus, caudate, putamen, brain stem, and cerebellum. One older patient progressed to develop multiple focal lesions in the cerebral and cerebellar cortex and subcortical white matter. The cause of deep and cortical gray matter injury in patients with SPLIS is not known with certainty. The appearance is not specific and resembles other toxic, metabolic, mitochondrial, infectious, and postinfectious disorders. The simultaneous presence of nephrotic syndrome with renal dysfunction, adrenal insufficiency, immunodeficiency, and sphingolipid deposition disease creates a complicated mix of possible pathophysiologic mechanisms of brain injury.
One pattern of injury affects the dopaminergic pathways, particularly the globus pallidus and dentate nucleus, but also the red nucleus and substantia nigra. These dopaminergic neural pathways are characterized by high rates of metabolism and mineral transport. Thus, they may be especially sensitive to changes in energy production, myelin composition, vascular supply, or the abnormal accumulation of cellular sphingolipids, which influences mitochondrial function and cell fate decisions in response to stress.13 The progressive nature of these changes has been shown on serial imaging. Routine and susceptibility-weighted imaging sequences have shown mineral deposition within or at the margin of lesions affecting the globus pallidus, thalamus, and dentate nucleus. Microhemorrhage and necrosis were suspected. However, analysis of phase images in 1 patient suggested the presence of calcium rather than iron or manganese. Disorders of calcium phosphate metabolism are known to lead to mineral deposition in the basal ganglia, particularly the globus pallidus; however, the deposition appears passive and less destructive than that observed in patients with SPLIS. Neurodegeneration with brain iron accumulation, including pantothenate kinase-associated neurodegeneration (PKAN), leads to iron deposition in the dopaminergic pathways. The appearance resembles that of some of our patients, but the presence of iron has not been proved in patients with genetically proved SPLIS. We recently had the opportunity to image and review many patients with PKAN as part of a novel drug-treatment trial.14 Other variants of neurodegeneration with brain iron accumulation were excluded. All patients had symmetric SWI signal changes in the globus pallidus, substantia nigra, red nucleus, and dentate nucleus. Some patients with SPLIS have involvement of these structures, which progresses to a much more robust disease than typical of PKAN. The presence of cortical and cerebellar gray and white matter involvement seems to distinguish SPLIS from PKAN in this regard.
The 1 patient whose fetal images we reviewed demonstrated adrenal enlargement and calcification. Among the known causes of adrenal calcification are Wolman disease and Niemann-Pick disease, both of which, like SPLIS, are disorders of lipid metabolism.
The molecular pathogenesis of SPLIS neuropathy is unknown. Mice constitutively lacking SGPL1 in brain neurons exhibit cognitive and motor deficits.15 SGPL1 disruption blocks autophagic flux and induces accumulation of aggregation-prone proteins in hippocampal neurons and brain tissue.15,16 S1P can act as a neurotoxin by inducing inappropriate elevations in cellular calcium, activating the unfolded protein response.17,18 In addition, sphingolipids such as S1P and ceramide exert effects on apoptosis, autophagy, exosome release, neuroinflammation, mitochondrial function, and the vasculature.19 Knockout mice lacking the ability to produce S1P or signal through S1P receptor 1 have interrupted angiogenesis and neurogenesis, are embryonic lethal, and exhibit neural tube closure defects and increased neuronal apoptosis.20
Due to the novelty of SPLIS and its diverse clinical presentations, patients with SPLIS may be challenging to recognize. Most are identified by next-generation sequencing studies of patients presenting with congenital or infantile nephrotic syndrome or nephrotic syndrome associated with other congenital anomalies. Some commercially available diagnostic panels for genetic testing of patients with these conditions include SGPL1, but this is not universal. Furthermore, gene panels focused on other disease characteristics have not yet incorporated SGPL1. Therefore, clinicians in all areas of pediatrics including radiologists who may encounter the MR imaging patterns described in this report should have a high index of clinical suspicion. Delay in diagnosis may lead to complications due to unrecognized conditions such as primary adrenal insufficiency or immunodeficiency. Early diagnosis would provide a window of opportunity for intervention before progression of clinical sequelae. Current treatments for SPLIS are largely supportive. However, targeted therapeutic strategies are in development and could potentially be leveraged to avert the devastating neurodegenerative component of the syndrome.12
Footnotes
This work was supported by Public Health Service grant R01DK115669 (J.D.S.), a grant from the Swim Across America Foundation (J.D.S.), and the National Institutes of Health grant R01 NIH-DK076683 (F.H.).
Disclosures: Nicole Weaver—UNRELATED: Grants/Grants Pending: National Institutes of Health, Comments: K08 award through the National Heart, Lung, and Blood Institute; Travel/Accommodations/Meeting Expenses Unrelated to Activities Listed: hotel accommodations for 2 nights at Costello Syndrome Family Network meeting in July 2019. Evren Gumus—UNRELATED: Employment: Muğla Sıtkı Koçman University. Friedhelm Hildebrandt—UNRELATED: Board Membership: Goldfinch Bio, Comments: cofounder and scientific advisory board member. Julie D. Saba—RELATED: Grant: National Institutes of Health and Swim Across America Foundation, Comments: This work was supported by a grant from the National Institutes of Health DK115669 to myself, and funds for research from the Swim Across America Foundation.* *Money paid to the institution.
Indicates open access to non-subscribers at www.ajnr.org
References
- Received March 4, 2020.
- Accepted after revision June 1, 2020.
- © 2020 by American Journal of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}