
Abstract
SUMMARY: Neuroradiologists play a key role in brain tumor diagnosis and management. Staying current with the latest classification systems and diagnostic markers is important to provide optimal patient care. Publication of the 2016 World Health Organization Classification of Tumors of the Central Nervous System introduced a paradigm shift in the diagnosis of CNS neoplasms. For the first time, both histologic features and genetic alterations were incorporated into the diagnostic framework, classifying and grading brain tumors. The newly published 2021 World Health Organization Classification of Tumors of the Central Nervous System, May 2021, 5th edition, has added even more molecular features and updated pathologic diagnoses. We present, summarize, and illustrate the most salient aspects of the new 5th edition. We have selected the key “must know” topics for practicing neuroradiologists.
ABBREVIATIONS:
- DGONC
- diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters
- EPN
- ependymoma
- ETMR
- embryonal tumor with multilayered rosettes
- FISH
- fluorescence in situ hybridization
- NEC
- not elsewhere classified; NOS = not otherwise specified
- MB
- medulloblastoma
- MGNT
- myxoid glioneuronal tumor
- MVNT
- multinodular and vacuolating neuronal tumor
- PF
- posterior fossa
- SC
- spinal cord
- ST
- supratentorial
- WHO
- World Health Organization
- IDH
- isocitrate dehydrogenase
Publication of the 2016 World Health Organization (WHO) Classification of Tumors of the Central Nervous System introduced a paradigm shift in the diagnosis of CNS neoplasms. For the first time, both histologic features and genetic alterations were incorporated into the diagnostic framework, classifying and grading brain tumors.
The rapidly evolving molecular landscape demanded interim updates between WHO editions (typically every 7 years). In late 2016, the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy (cIMPACT-NOW) was created under the sponsorship of the International Society of Neuropathology to provide such updates.1,2 To date, 7 updates3⇓⇓⇓⇓⇓⇓-10 have been published to bridge the gap between the 4th edition and the newly published (May, 2021) 5th edition of the famed “blue book.”11
We present, summarize, and illustrate the most salient aspects of the new 5th edition. We have selected the key “must know” topics for practicing neuroradiologists. The 2021 WHO Classification of Tumors of the Central Nervous System can be ordered in either print or digital form from the WHO website and should be part of every neuroradiologist’s library.
General Features and Recommendations
Tumor Taxonomy and Nomenclature.
Prior editions used the terms “entities” and “variants.” The current edition uses the terms “types” and “subtypes” and keeps tumor names as simple as possible. Newly recognized or redefined types and subtypes are summarized in the Online Supplemental Data.
Tumor Grading.
The 5th edition uses Arabic numerals instead of Roman numerals to conform to other WHO grading systems and decrease the likelihood of typographic errors when grading within types. Tumor grades are now designated specifically as CNS WHO grades 1–4 (“CNS” is always added to distinguish the grading system from those of systemic neoplasms because CNS grading differs conceptually, eg, grading of diffuse astrocytomas from 2 to 4, without a 1).
Not Otherwise Specified and Not Elsewhere Classified.
Not otherwise specified (NOS) is used when molecular information is not available/not performed/not successful. Not elsewhere classified (NEC) is used when necessary diagnostic testing was successfully performed but the results do not readily permit a WHO diagnosis (eg, entities that are not yet recognized as part of the WHO Classification).3 NOS and NEC can be used for any tumor type.
“Layered” Reports and Integrated Diagnosis.
A matrix approach to an integrated pathologic diagnosis is used throughout the 5th edition (Table). Features such as location (eg, cerebrum or cerebellum), histopathology, and molecular information (when available) are combined into the top layer (in reality the “bottom line”) to create an integrated diagnosis. Tumor grade reflects a combination of histologic features and genetically defined mutation status. If molecular information is unavailable, tumor entities are generally designated by NOS.
Layered neuropathology diagnosisa
General Taxonomy
The WHO 5th edition organizes CNS neoplasms into several major groups: gliomas, glioneuronal, and neuronal tumors; choroid plexus tumors; embryonal tumors; pineal tumors; cranial and paraspinal nerve tumors; meningioma; mesenchymal, nonmeningothelial tumors; melanocytic tumors; hematolymphoid tumors; germ cell tumors; tumors of the sellar region; and metastases to the CNS. In this overview, we will focus on the tumor groups with specific changes such as newly recognized tumor entities, revised nomenclature, and restructured tumor groupings.
Gliomas, Glioneuronal, and Neuronal Tumors
Gliomas, glioneuronal, and neuronal tumors, along with the embryonal tumors, have undergone the most important changes since the 2016 4th edition. There are now 14 newly recognized (“new”) gliomas and glioneuronal tumors in the 5th edition of the blue book. In addition, for the first time, the WHO classification divides diffuse gliomas into adult-type and pediatric-type neoplasms.
Gliomas
Neuropathologic Essentials.
Glioma characterization requires more than simply determining whether a tumor exhibits 1p/19q codeletion on fluorescence in situ hybridization (FISH) and is isocitrate dehydrogenase (IDH) mutant or IDH-wildtype on immunohistochemistry, to implement the 2021 WHO classification fully. For example, IDH-wildtype diffuse astrocytic gliomas in patients 55 years of age and younger should also be investigated for noncanonical (ie, non-R132H) IDH1 mutations and IDH2 mutations. In other molecular markers such as loss of ATRX expression or TERT promoter mutations, the presence of TP53 or histone H3 mutations, EGFR amplification, or CDKN2A/B alterations, and so forth need to be evaluated in specific diagnostic pathways.
Some genetic changes have convenient immunohistochemistry surrogate assays (eg, IDH1 R132H, ATRX, p53, BRAF V600E, H3K27M, H3 G34R/V), while others can be detected with FISH (eg, CDKN2A/B homozygous deletion, EGFR amplification, 1p/19q codeletion). Next-generation sequencing assays will detect many of these and other events such as mutations and fusions. Methylome profiling has also emerged as a powerful tool that can be used itself for classification and can also either directly or indirectly identify many of the above molecular alterations.
Four general groups of diffuse gliomas are recognized in the 2021 WHO classification: 1) adult-type diffuse gliomas, 2) pediatric-type diffuse low-grade gliomas, 3) pediatric-type diffuse high-grade gliomas, and 4) circumscribed astrocytic gliomas.
Adult-type diffuse gliomas are astrocytoma, IDH-mutant; oligodendroglioma, IDH-mutant and 1p/19q-codeleted; and glioblastoma, IDH-wildtype. IDH-mutant diffuse astrocytomas are now graded 2–4 within type; the terms IDH-mutant “anaplastic astrocytoma” and “glioblastoma” have been dropped. In addition, if an IDH-mutant diffuse astrocytoma exhibits CDKN2A/B homozygous deletion, it is designated as a CNS WHO grade 4 neoplasm, even if histologic features of malignancy such as necrosis and microvascular proliferation are absent.
Imaging features suggestive of an IDH-mutant diffuse astrocytoma grade 2 include a homogeneous T2-hyperintense circumscribed supratentorial mass typically in the frontal or temporal lobes without calcification or enhancement. The T2-FLAIR mismatch sign, characterized by T2 homogeneity of the mass with relatively hypointense signal throughout most of the lesion on FLAIR compared with T2 sequences except for a peripheral rim of hyperintense signal, is highly predictive of IDH-mutant diffuse astrocytoma. The T2-FLAIR mismatch sign has high specificity but low sensitivity for IDH-mutant diffuse astrocytomas.12,13 Imaging features of IDH-mutant diffuse astrocytoma grade 3 may be indistinguishable from grade 2 IDH-mutant diffuse astrocytomas. However, grade 3 astrocytomas may have T2 heterogeneity and enhancement as well as elevated maximum relative CBV. The mean maximum relative CBV is significantly higher in WHO grade 3 astrocytomas than in WHO grade 2 astrocytomas.14 Imaging features typical of oligodendroglioma, IDH-mutant and 1p/19q-codeleted, tumors include frontal lobe location, heterogeneity, and calcification with variable enhancement (Fig 1).12

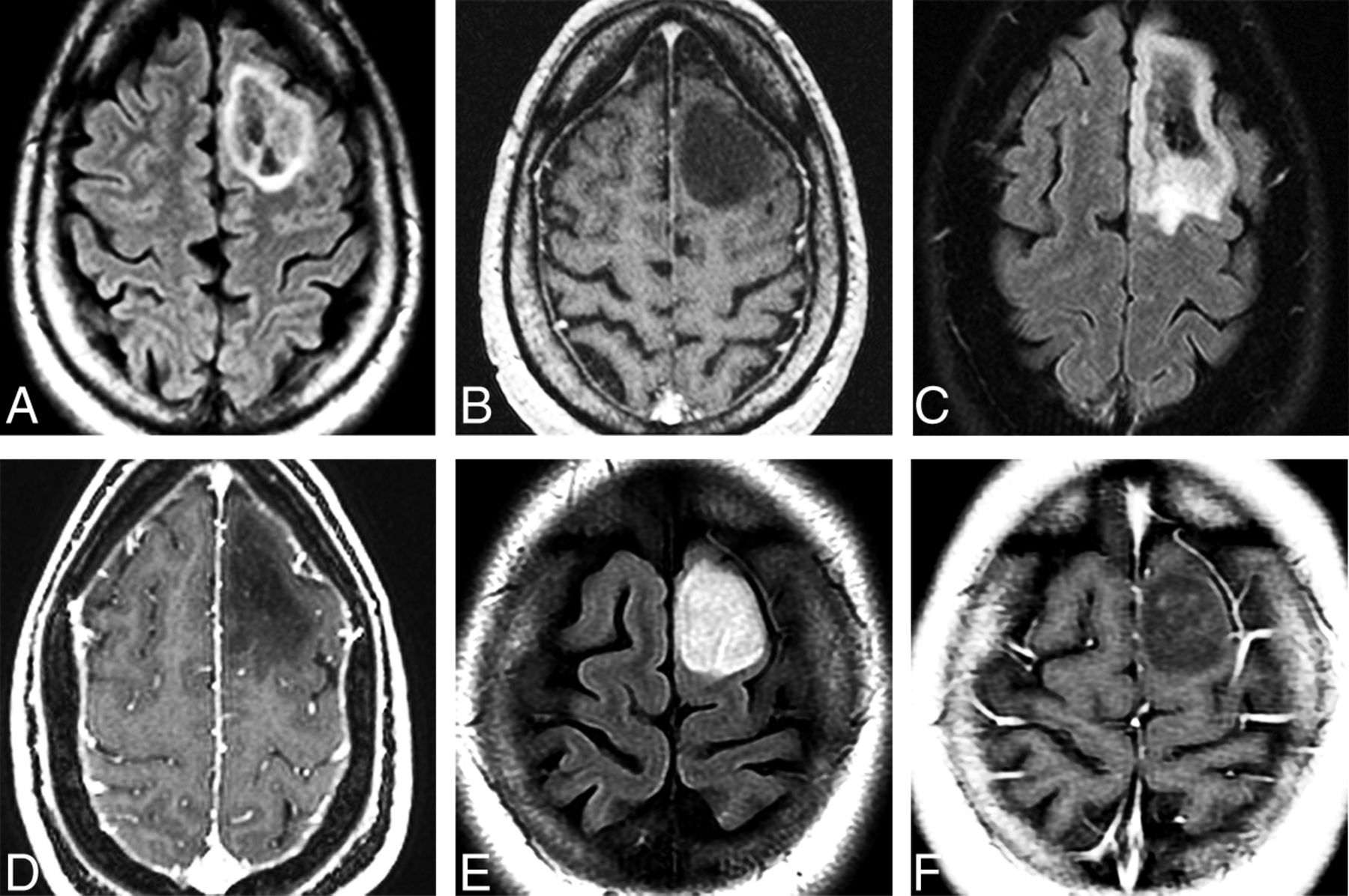
Adult-type diffuse gliomas. Series of 3 cases illustrates the importance of complete IDH mutation status determination and the investigation of other molecular markers in evaluation of adult-type diffuse astrocytomas. Axial FLAIR (A) and postcontrast T1WI (B) in a 54-year-old man with a first-time seizure shows a well-delineated left frontal lobe mass with a hyperintense rim surrounding a mixed signal mass. No enhancement is present. Pathology disclosed diffuse astrocytoma without necrosis or microvascular proliferation. Immunohistochemistry demonstrated that the tumor was IDH-mutant. Next generation sequencing disclosed CDKN2A/B homozygous loss, so the tumor was upgraded to WHO CNS grade 4. Axial FLAIR (C) and postcontrast T1WI (D) in a 44-year-old woman with a first-time seizure demonstrate a left frontal mass that was completely resected. Pathology findings were consistent with WHO CNS grade 3. Initial immunohistochemistry was negative for IDH1 mutation, but further investigation disclosed the presence of an IDH2 mutation. Final pathologic diagnosis is diffuse astrocytoma, IDH-mutant, grade 3. The patient is alive without evidence of disease 4 years after the initial diagnosis. Axial FLAIR (E) and postcontrast T1WI (F) in a 24-year-old woman with a first-time seizure show a well-delineated nonenhancing left frontal lobe mass that was surgically resected. Histologically, the tumor was WHO CNS grade 2 but IDH-wildtype on immunohistochemistry. No further investigation was conducted. One year later, the tumor recurred and re-resection demonstrated EGFR amplification and was, therefore, upgraded to glioblastoma (WHO CNS grade 4). The patient died of disseminated disease 18 months after the initial diagnosis.
The presence of any one of the following 5 criteria is sufficient to designate an IDH-wildtype diffuse astrocytic glioma as a glioblastoma. IDH-wildtype is characterized by the following: microvascular proliferation or necrosis or TERT promotor mutation or EGFR gene amplification or +7/–10 chromosome copy number changes. Such tumors are no longer called “diffuse astrocytic glioma, IDH-wildtype with molecular features of glioblastoma multiforme.” If an IDH-wildtype tumor exhibits none of these histologic or molecular features (eg, appears as a lower grade than a glioblastoma, CNS WHO grade 4), it would be classified as diffuse astrocytoma, NEC (Fig 1).
Pediatric-type diffuse low-grade gliomas are diffuse astrocytomas, MYB or MYBL1-altered; angiocentric gliomas (Fig 2); polymorphous low-grade neuroepithelial tumor of the young (a newly recognized entity exhibiting oligodendroglioma-like histology with variable morphology and MAPK-pathway alterations);15 and diffuse low-grade gliomas, MAPK pathway–altered. Angiocentric gliomas are T2-hyperintense masses typically in the temporal or frontal lobe cortex in young patients with seizures (Fig 2). Polymorphous low-grade neuroepithelial tumors of the young are typically well-circumscribed T2-hyperintense lesions on MR imaging with central calcification and peripheral cystic components (Fig 3). They are commonly supratentorial, most often within the temporal lobe.15,16 Diffuse low-grade glioma, MAPK pathway–altered, is a group of neoplasms that are IDH- and H3-wildtype and include most tectal gliomas. Up-regulation of the RAS/MAPK pathway is almost universal in these lesions, with a spectrum of FGFR1 and BRAF mutations. Histologic features of malignancy and molecular alterations such as CDKN2A/B mutations are absent.17,18 The classic tectal gliomas are not considered a distinct WHO entity. Most fit histologically and genetically into either pilocytic astrocytoma with BRAF alterations and NRAS mutations or diffuse low-grade glioma, MAPK pathway–altered.

Pediatric-type diffuse low-grade glioma. Axial T2 (A) MR image in a 7-year-old boy with a diffuse astrocytoma, MYB-altered, shows a hyperintense mass in the pons with no significant surrounding edema. There was no enhancement and no diffusion restriction of the mass (not shown). Axial FLAIR (B), postcontrast T1 (C), and arterial spin-labeling (ASL) (D) in a 1-year-old child with an angiocentric glioma show a FLAIR hyperintense mass involving the cortex and subcortical white matter of the left frontal lobe. There is no enhancement (C) and decreased perfusion (D) on ASL imaging.

Two patients with the WHO 2021 new-entity polymorphous low-grade neuroepithelial tumor of the young (PLNTY). Axial FLAIR (A) and postcontrast T1-weighted (B) MR images in a 19-year-old man with refractory epilepsy show a hyperintense, nonenhancing mass in the cortex and subcortical white matter of the left temporal lobe. Axial FLAIR (C) and susceptibility-weighted (D) MR images and a noncontrast CT image (E) in a 19-year-old woman with progressive seizure show a FLAIR-hyperintense, SWI-hypointense mass with characteristic calcification seen on CT in the right medial temporal lobe (Case courtesy of M. Castillo, MD).
Pediatric-type diffuse high-grade gliomas are defined primarily by molecular features and include diffuse midline glioma, H3K27-altered (note that the term “mutant” has been changed) (Fig 4); diffuse hemispheric glioma, H3 G34-mutant (an H3F3A-mutant, IDH-wildtype tumor that exhibits glioblastoma-like histology, often with primitive embryonal regions) (Fig 5); diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype (a group of tumors with different possible genotypes); and infant-type hemispheric glioma (Fig 6).19,20 The classic diffuse intrinsic pontine gliomas seen on MR imaging as expansile T2-hyperintense lesions are most commonly diffuse midline gliomas, H3K27-altered pathologically, similar to the 2016 WHO description. However, other gliomas may affect the pons.21 In addition to the more common pediatric brainstem glioma presentation, H3K27-altered high-grade gliomas occur in adults and have the same lethality as in their pediatric counterparts.18 Unilateral thalamic or bithalamic lesions are common in H3K27-altered high-grade gliomas as is aggressive local spread and early metastatic dissemination.

Pediatric-type diffuse high-grade gliomas. Diffuse midline glioma, H3K27-altered in an 8-year-old girl with cranial neuropathies. Axial T2 (A) and FLAIR (B) MR images show an expansile, hyperintense pontine mass. Axial postcontrast T1 MR image (C) shows heterogeneous enhancement within the mass. Arterial spin-labeling (ASL) perfusion (D) shows increased perfusion. E, Axial FLAIR MR image shows a bithalamic hyperintense mass. Postcontrast T1WI showed no significant enhancement, and ASL perfusion showed increased perfusion within the bilateral thalami (not shown). These WHO grade 4 tumors have a poor prognosis.

Diffuse hemispheric glioma, H3 G34-mutant and IDH-wildtype tumor in an 8-year-old boy. A, Axial FLAIR shows a large, very heterogeneous right temporal lobe mass with minimal surrounding edema. B, An ADC map in the same case shows restricted diffusion consistent with a high-cellularity tumor. C, Arterial spin-labeling perfusion shows decreased perfusion in the tumor. In pediatric tumors, perfusion is often less helpful compared with diffusion-weighted imaging in discriminating tumor grade. Histology demonstrated necrosis, hemorrhage, and neovascularity in a glioblastoma-like pattern, consistent with grade 4 tumor.

Infant-type hemispheric glioma, NOS. A male neonate child with macrocephaly and bulging fontanelles had a large, heterogeneous-appearing mass on an emergent CT scan (not shown). Axial T2-weighted (A) and postcontrast T1-weighted (B) MR images show a very heterogeneous mass with enhancement involving almost the entirety of the left cerebral hemisphere.
Infant-type hemispheric gliomas are tumors of early childhood that exhibit high-grade astrocytic (often glioblastoma-like) histologic features with alterations in ALK/ROS1/NTRK/MET. A large, bulky nearly holohemispheric, heterogeneous-appearing tumor with intratumoral hemorrhage is typical.
Circumscribed astrocytic gliomas include long-recognized neoplasms (such as pilocytic and subependymal giant cell astrocytomas) and 2 new entities, high-grade astrocytomas with piloid features and astroblastoma, MN1-altered. While not designated as separate entities, the molecular characterization of low-grade gliomas has had a profound effect on their treatment. For instance, the identification of BRAF V600E mutations allows targeted disruption by using BRAF inhibitors, with favorable clinical results.22
The diagnosis of high-grade astrocytoma with piloid features recognizes unusual cases in which a relatively circumscribed tumor with distinct piloid cytology occurs in the setting of a more malignant astrocytoma (WHO grades 3 or 4).23 These tumors usually occur in adults, exhibit CDKN2A/B deletions, and have a distinct DNA methylation profile that differs from the typical childhood pilocytic astrocytomas. Most of these tumors occur in the posterior fossa (PF), are T2-hyperintense, and show heterogeneous enhancement.23 The relationship with so-called “anaplastic pilocytic astrocytomas” and pre-existing pilocytic astrocytomas is currently undetermined.
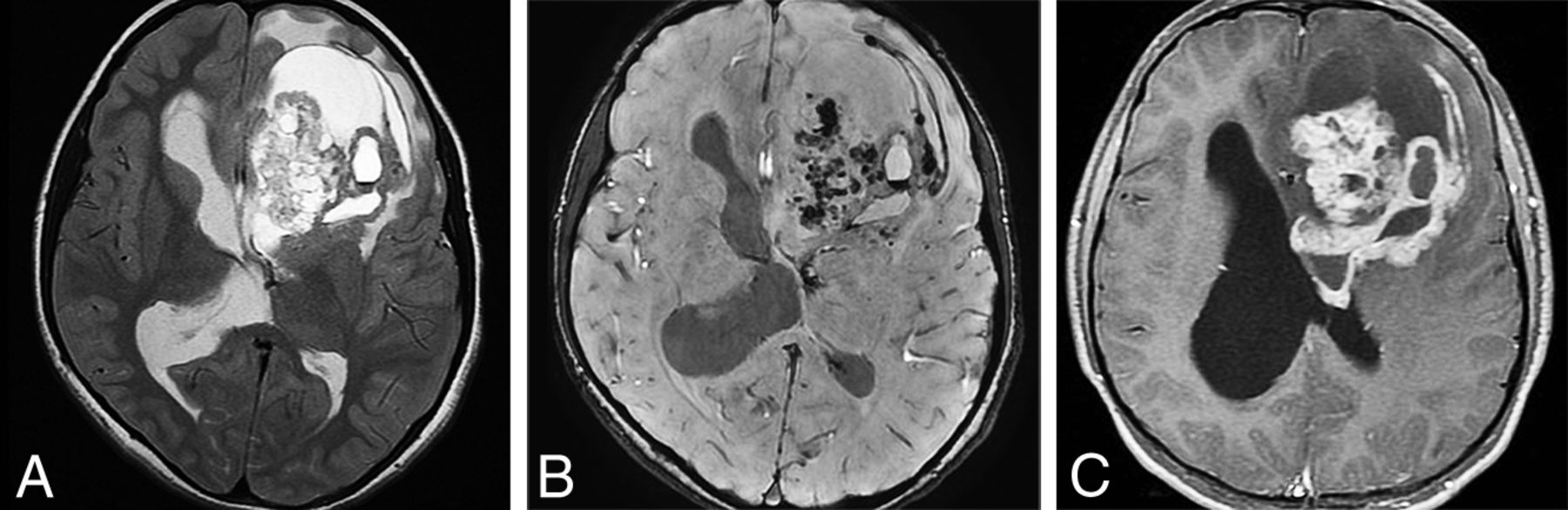
Astroblastoma, MN1-altered, is newly classified as a circumscribed astrocytic glioma (in 2016 it was categorized with “other gliomas”). MN-1 alterations are present in 70%.24 If MN-1 alteration is absent or not determined, the tumor is designated NEC or NOS, respectively. Most astroblastomas are located superficially in a cerebral hemisphere and are relatively well-circumscribed tumors that can be multicystic or “bubbly” in appearance. Edema is minimal or absent (Fig 7).8,25,26 No formal grade for astroblastoma is assigned in the 5th edition.

Axial T2WI in a 19-month-old child with astroblastoma, MN1-altered. A, Axial T2WI shows a bubbly-appearing mixed-signal hemispheric mass with little surrounding edema. B, Postcontrast T1WI shows that the mass enhances strongly but heterogeneously.
Miscellaneous 5th Edition Glioma Items.
In 2021, pilomyxoid astrocytoma continues to be considered a variant of pilocytic astrocytoma, not a distinct entity. The location modifier (third ventricle) has been dropped from choroid glioma. Like medulloblastoma, it only occurs in 1 location; therefore, a location modifier is not necessary.
Glioneuronal and Neuronal Tumors
Ganglioglioma, desmoplastic infantile ganglioglioma/astrocytoma, dysembryoplastic neuroepithelial tumor, and other mixed glioneuronal tumors such as rosette-forming glioneuronal tumor are unchanged. Newly clarified and added tumor entities include diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters (DGONC), myxoid glioneuronal tumor (MGNT), and multinodular and vacuolating neuronal tumor (MVNT).
DGONC is included in the 5th edition as a provisional entity, defined primarily by a DNA methylation profile. As the name implies, histology is oligodendroglioma-like with large cells that have clusters of nuclei. DGONCs are primarily pediatric tumors but can occur at all ages.27
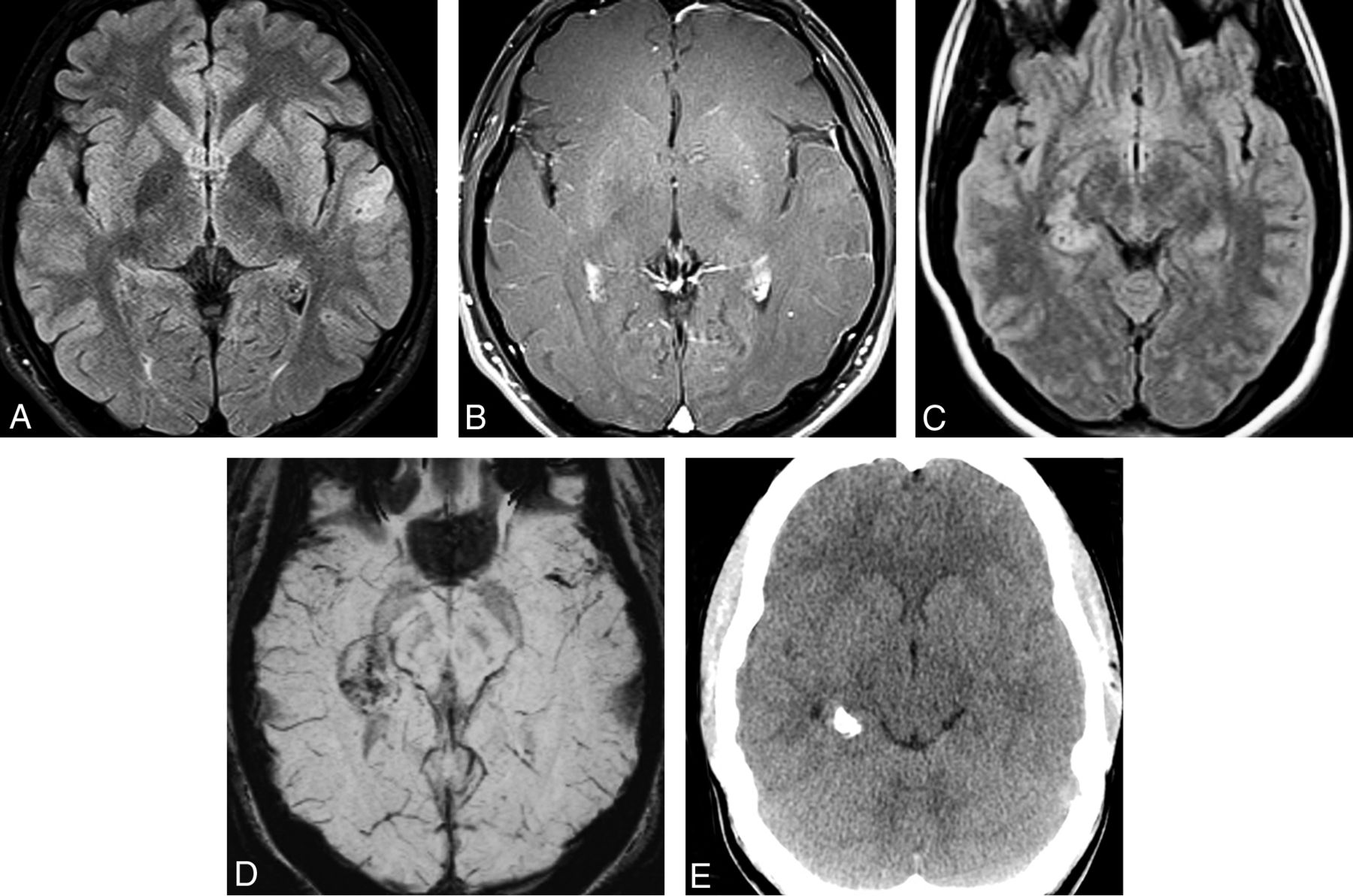
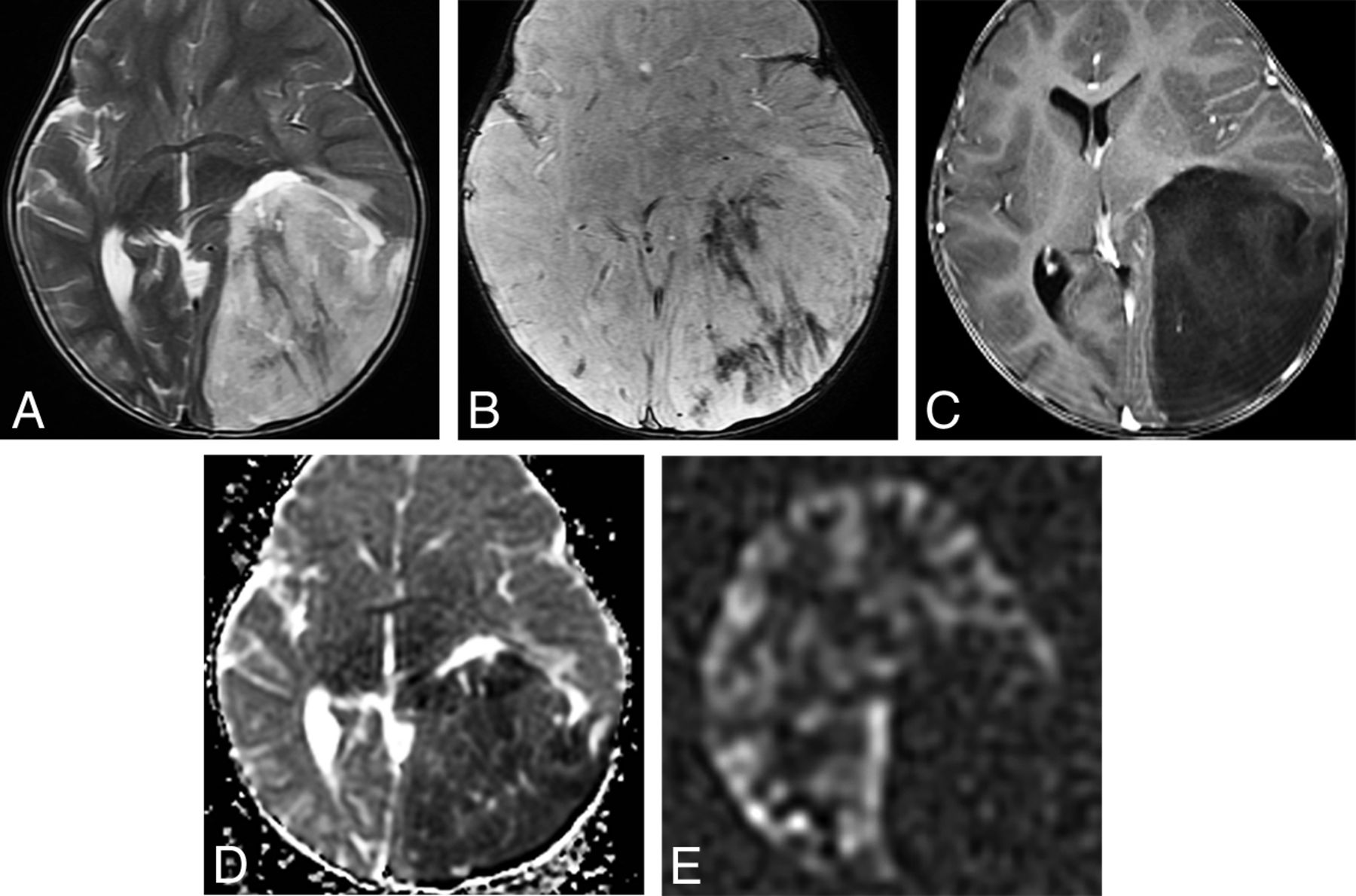
MGNT is a CNS WHO grade 1 neoplasm that is stereotypically located in the septum pellucidum, though it can also occur in the corpus callosum and periventricular white matter. Oligodendrocyte-like tumor cells are embedded in a prominent myxoid stroma. Specific mutations in PDGFRA are definitional.28,29 In addition to location, suggestive imaging findings include T2 hyperintensity, peripheral FLAIR hyperintensity, and lack of enhancement (Fig 8). MGNTs are considered CNS WHO grade 1 neoplasms, but many cases exhibit ventricular dissemination or local recurrence/progression.8,29

Two cases of MGNT are illustrated. A, Axial T2WI in a 14-year-old boy shows an extremely hyperintense, slightly bubbly mass in the left juxtaventricular white matter. B, Axial FLAIR shows a hyperintense rim surrounding a largely isointense center of the mass. Smaller-but-similar-appearing lesions are adjacent to the mass. The mass did not enhance following contrast administration. C, Sagittal T1WI in a 39-year-old man shows a well-demarcated mass in the corpus callosum rostrum/septum pellucidum. D, The mass is extremely hyperintense on T2WI. E, FLAIR shows that the mass has a hyperintense rim with an isointense center. The mass is thought to represent an MGNT because of its classic location and signal characteristics but is not biopsy-proven.
MVNT was considered a pattern of ganglion cell tumors in the 2016 WHO. Whether MVNT represented a neoplastic or malformative process was then unknown. Now MVNTs are recognized as clonal neoplasms of the MAPK pathway with mutations in MAPK2K1 and BRAF (excluding V600E) as well as FGFR2 fusions. MVNTs are nonprogressive CNS WHO grade 1 lesions. MR imaging features are virtually pathognomonic with clusters of T2-FLAIR hyperintense nodules (little bubbles) along the undersurface of the cerebral cortex and subcortical white matter.30,31 MVNT-like lesions have also been reported in the posterior fossa.32
Ependymal Tumors
Ependymomas (EPNs) are the last of the glioma/glioneuronal/neuronal tumor groupings. The 2016 WHO divided ependymal tumors into 4 subgroups: subependymoma (CNS WHO grade I), myxopapillary ependymoma (CNS WHO grade I), ependymoma (CNS WHO grade II), and anaplastic ependymoma (CNS WHO grade III).
In a major departure since the 2016 WHO, ependymomas are now uniquely grouped by location.8,9 The WHO recognized 3 distinct anatomic sites: supratentorial (ST), PF, and spinal cord (SC) EPNs. Within each specific anatomic site, molecularly defined subgroups are defined by gene and DNA methylation profiling. Each differs in location, age, prognosis, and clinicopathologic characteristics.9
ST-EPN.
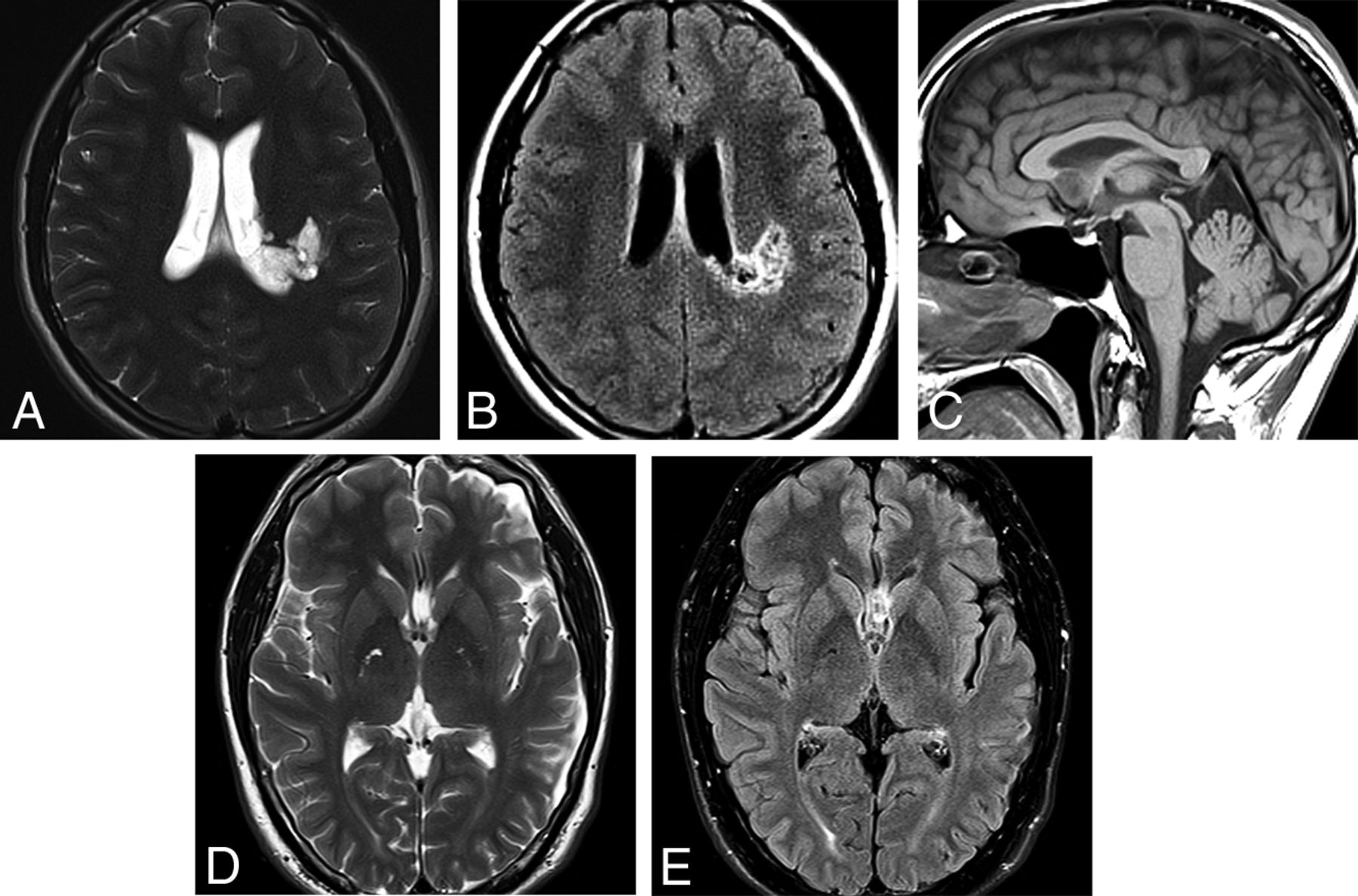
ZFTA fusion–positive ependymomas (formerly RELA-fusion ependymoma) are extraventricular hemispheric tumors that exhibit rearrangement of partners with the ZFTA (formerly C11orf95) genes (Fig 9). These tumors are the largest group of currently defined ST-EPNs. They occur in both children and adults and are designated CNS WHO grade 2 or 3 neoplasms. They appear as relatively well-defined mixed cystic-solid masses on imaging studies. YAP1-fusion ST-EPNs are found mostly in children younger than 3 years of age and have a better prognosis than ZFTA ependymomas.33 Tumors that do not have the ZFTA- or YAP1-fusion events are termed ST-EPN are described by their histologic features.

ZFTA fusion–positive ependymoma in an 11-year-old girl. A, Axial T2WI shows a large, bulky, heterogeneous left frontal mass. B, Susceptibility-weighted scan shows intratumoral hemorrhage. C, Strong-but-very heterogeneous enhancement is seen on postcontrast T1WI.
PF-EPNs can now be divided molecularly into 2 subgroups: PF-EPN A and PF-EPN-B.34 PF-A ependymomas occur mainly in infants, exhibit loss of H3K27me3 expression on immunohistochemistry, exhibit EZHIP overexpression, and have significantly worse outcome than PF-EPN-B tumors. PF-EPN-B tumors are more common in older children and adults. Posterior fossa ependymomas are characterized on MR imaging as a lobulated, heterogeneous mass in the body or inferior fourth ventricle, which often extends through the foramen of Magendie into the cisterna magna or through the foramina of Luschka into the cerebellopontine angle cisterns. Calcification and cystic changes are often seen. Both the histology and imaging features of the 2 posterior fossa ependymoma subgroups are similar, but PF-EPN-A tumors are more likely to have a lateral location within the posterior fossa and show cerebellar invasion.9,35 Tumors that cannot be evaluated further are termed posterior fossa ependymomas and are described by their histologic features.
Spinal Ependymomas.
The 2021 WHO recognizes a new type of spinal cord ependymoma with MYCN–amplification. MYCN-amplified ependymoma is mostly found in adults and exhibits anaplastic histology. These tumors are typically located in the cervical or thoracic spinal cord and extend over many spinal segments. These spinal cord tumors are heterogeneously T2-hyperintense and enhancing and are typically extramedullary or have an exophytic portion if intramedullary and are characterized by leptomeningeal disease. Early dissemination and poor prognosis are typical.9,36 Of note, myxopapillary ependymomas are now designated CNS WHO grade 2 neoplasms because their biologic behavior is more consistent with this designation.9
Choroid Plexus Tumors
The classification of choroid plexus tumors remains unchanged in 2021, though these are now listed separately from the glial and glioneuronal neoplasms.
Embryonal Tumors
The 2021 WHO classifies CNS embryonal tumors into 2 groups: medulloblastoma and other CNS embryonal tumors (the term “primitive neuroectodermal tumor” has been abandoned since 2016).
Medulloblastoma
As in 2016, medulloblastomas (MBs) can be either molecularly or histologically defined. The molecularly-defined MB subgroups are defined by DNA methylation or transcriptome profiling and remain unchanged: medulloblastoma, WNT-activated; medulloblastoma, SHH-activated and TP53 wild-type; medulloblastoma, SHH-activated and TP53-mutant; and medulloblastoma, non-WNT/non-SHH.11,37-39
Medulloblastoma, WNT-activated, represents approximately 10% of MBs. There are 2 age-determined subtypes: children and adults. WNT MBs can be found in all posterior fossa locations and are thought to arise from the lower rhombic lip. Imaging studies suggest that the cerebellar peduncle and cerebellopontine angle are the most characteristic but not the only location. WNT-activated MBs have the best prognosis of all 4 groups. Metastases are rare at diagnosis, and the 5-year survival rate is 95%.11,37,38
Medulloblastoma, SHH-activated/TP53 wild-type, represents approximately 30% of MBs overall but accounts for nearly two-thirds of MBs occurring between 3–16 years of age. This MB subgroup has the most striking biologic, pathologic, and clinical heterogeneity of all 4 subgroups. SHH-activated MBs arise from granule neuron progenitor cells in the upper rhombic lip, so a cerebellar hemispheric location is typical. These MBs have 4 provisional molecular subtypes as defined by DNA methylation or transcriptome profiling: SHH-1 and SHH-2 are the most common subgroups to exhibit desmoplastic or medulloblastoma with extensive nodularity histology. Desmoplastic MBs are more common in adults and have a predilection for the lateral cerebellar hemisphere.37 SHH-3 and SHH-4 most commonly exhibit classic or large-cell anaplastic histology and can be found in all locations.39,40
Medulloblastoma, SHH-activated/TP53-mutant, is the rarest of the MB subtypes and has the worst overall prognosis.11,40
Medulloblastoma, non-WNT/non-SHH, is the most common MB subtype, representing 50%–60% of all MBs. This subtype encompasses the former group 3 (20%) and group 4 (40%–50%) MBs. This subtype has 8 molecular subgroups (Gp3/4–1 to Gp3/4–8) as determined by methylation profiling. Non-SHH, non-WNT MBs can be found in all locations and often exhibit minimal or no enhancement.11
Other CNS Embryonal Tumors
This group of “other” embryonal tumors includes atypical teratoid/rhabdoid tumor and the addition of several “new” tumor types: a provisional type called cribriform neuroepithelial tumor and CNS tumor with BCOR internal tandem duplication. One embryonal tumor with a newly identified genotype is CNS neuroblastoma, FOXR2-activated. This group also includes embryonal tumor with multilayered rosettes (ETMR).
Cribriform Neuroepithelial Tumor.
Cribriform neuroepithelial tumor (provisional diagnosis) is a nonrhabdoid neuroectodermal tumor characterized molecularly by loss of nuclear SMARCB1/INI1 expression and histologically by cribriform strands/ribbons. Cribriform neuroepithelial tumors occur near the ventricles in young children and have a better prognosis than atypical teratoid/rhabdoid tumors.40
CNS Tumor with BCOR Internal Tandem Duplication.
CNS tumors with BCOR internal tandem duplication are mostly hemispheric malignant tumors of children and adolescents that are characterized by internal tandem duplication in the BCOR gene, similar to other systemic tumors.41
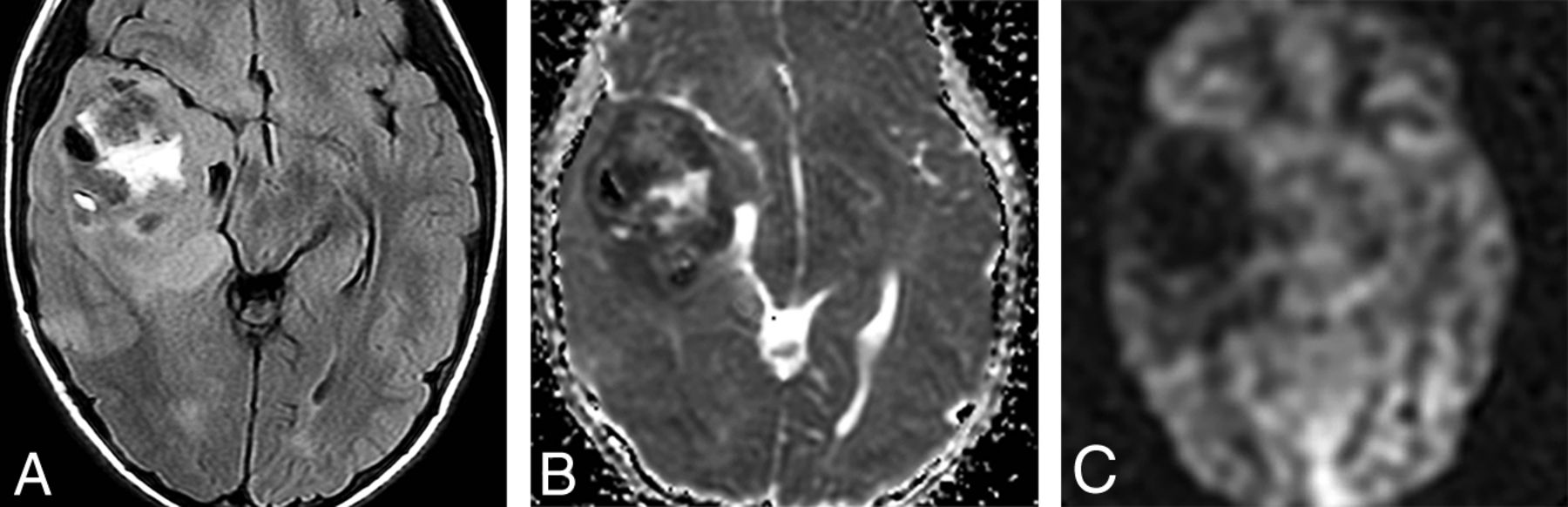
ETMR was included in 2016 specifically as chromosome 19 microRNA cluster–altered. An additional subtype, DICER1-mutated ETMR, has been recently described. ETMRs subsume many prior entities such as embryonal tumor with abundant neuropil and true rosettes, medulloepithelioma, ependymoblastoma, and many tumors formerly known as CNS primitive neuroectodermal tumors. ETMRs are tumors of infants and children younger than 4 years of age. They are seen on imaging studies as large, cellular, relatively well-demarcated-but-heterogeneous-appearing masses.42 While they do occur in the posterior fossa, most are supratentorial hemispheric lesions (70% of cases). Necrosis and intratumoral hemorrhage are common. Solid components of the tumors typically exhibit restricted diffusion. Enhancement varies from patchy, sparse to mostly absent (Fig 10).42

Embryonal tumor with multilayered rosettes in a 1-year-old girl. A, An axial T2-weighted scan shows a large, left parieto-occipital mass with little surrounding edema. B, The mass exhibits hemorrhage on susceptibility-weighted imaging and no enhancement following contrast administration (C). D, Strikingly restricted diffusion is seen on the ADC map. E, Arterial spin-labeling perfusion shows decreased perfusion in the tumor.
CNS Neuroblastoma, FOXR2-Activated.
CNS neuroblastoma is a newly recognized embryonal tumor that has a characteristic histology and FOXR2 gene alterations.43 These primary CNS neuroblastomas have a peak at 5 years of age and are characterized by neuronal differentiation, high vascularity, necrosis, and endothelial proliferation. Imaging shows a large, heterogeneous supratentorial mass with prominent cysts, necrosis, little surrounding edema, and variable enhancement.
Keep in mind that the imaging differential diagnosis of a large, bulky, heterogeneous hemispheric mass in an infant or young child includes ETMR, infant-type hemispheric glioma, ZFTA ependymoma, CNS neuroblastoma, FOXR2-activated and CNS embryonal tumor, NOS or NEC. The term primitive neuroectodermal tumor has been abandoned since 2016.
Pineal Tumors
With 1 exception, pineal tumors remain unchanged since 2016. A newly codified tumor, desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant, is now recognized. This rare tumor of the pineal region (not specifically the pineal gland) has both desmoplastic and myxoid changes but no histopathologic signs of malignancy.44 Only a limited number of cases have been reported with an age range of 15–61 years (median, 40 years).11
Cranial and Paraspinal Nerve Tumors
The term malignant melanotic nerve sheath tumor, previously called melanotic schwannoma, has been changed, in part, because it behaves more aggressively than nonmelanotic schwannomas and also to conform with soft-tissue nomenclature.
Meningiomas
In terms of the overall classification, the meningioma tumor group remains unchanged. However, there are a number of molecular alterations that are now recognized as diagnostically and prognostically useful.
Mesenchymal, Nonmeningothelial Tumors
Mesenchymal, nonmeningothelial tumors are divided into 2 groups: soft-tissue tumors and chondro-osseous tumors. The only major changes in 2021 are with soft-tissue tumors.
Soft-Tissue Tumors
Soft-tissue tumors are subcategorized into fibroblastic and myofibroblastic tumors, vascular tumors, skeletal muscle tumors, and tumors of uncertain differentiation. The term hemangiopericytoma is now considered obsolete, and the preferred term “solitary fibrous tumor” is used to correspond to extracranial solitary, fibrous tumors. Solitary, fibrous tumors are the most common nonmeningothelial mesenchymal neoplasm and share the common molecular feature of NAB2-STAT6 gene fusions. Tumor grades vary from WHO 1–3 (WHO grade III solitary fibrous tumors were previously referred to as “anaplastic hemangiopericytomas”). Imaging features often resemble those of meningiomas.
There are 3 newly recognized intracranial soft-tissue tumors: intracranial mesenchymal tumor, FET-creB fusion-positive; CIC-rearranged sarcoma; and primary intracranial sarcoma, DICER1-mutant.11,45
Intracranial mesenchymal tumor, FET-creB fusion-positive, often features specific EWSR1-creB1 fusions. These tumors can be extra-axial or intraventricular. The cerebral convexities are the most common location. They are typically T2-FLAIR hyperintense, exhibit strong enhancement, and often have a dural “tail.” The major differential diagnosis is atypical or anaplastic meningioma.11,45
CIC-rearranged sarcoma corresponds to similar soft-tissue tumors. Multiple CIC-fusion partners have been identified. Round-cell sarcomas with myxoid features are typical. These tumors can occur at any age but are most common in adolescents and young adults. They are highly aggressive and are designated as WHO CNS grade 4 lesions.11,45
Primary intracranial sarcoma, DICER1-mutant, is a highly-malignant CNS sarcoma that is part of the expanding spectrum of DICER1 and type 1 neurofibromatosis syndromes. This intracranial sarcoma primarily occurs in children and young adults, exhibiting malignant spindle cell morphology often with focal rhabdomyoblastic differentiation.11,45
Hematolymphoid Tumors
Other than grouping lymphomas and histiocytic tumors together as hematolymphoid tumors, no significant changes occurred in the 2021 WHO.
Germ Cell Tumors
No significant 2021 changes were made in germ cell neoplasms.
Tumors of the Sellar Region
Pituitary adenomas are now designated as pituitary adenoma/pituitary neuroendocrine tumors to correspond to systemic neuroendocrine tumors. Pituitary neuroendocrine tumors are now also classified according to adenohypophyseal cell lineages, rather than just by the hormone produced. Pituicytoma, granular cell tumor of the sellar region and spindle cell oncocytoma remain unchanged from WHO 2016, and though they are classified as separate tumor types, they are considered a related group of tumor types with possibly morphologic variations of the same tumor.11,46
One new tumor, pituitary blastoma, has been added to the 2021 WHO Classification of sellar region tumors. Pituitary blastomas are rare embryonal sellar neoplasms of infants (median age, 8 months) that are associated with somatic or germline DICER1 mutations. Pituitary blastomas are hypophyseal tumors that resemble a 10- to 12-week embryonic-stage pituitary gland. Primitive blastemal cells similar to those in pleuropulmonary blastomas, neuroendocrine cells, and Rathke-type epithelium in rosettes/glandular structures are characteristic. Pituitary blastomas are designated WHO CNS grade 4 neoplasms.11,45
Summary
The 2021 5th edition WHO Classification of CNS neoplasms (the series popularly known as the Blue Books) builds on the trend of molecular tumor classification first introduced in the 2016 (4-plus) edition. Gliomas are divided into adult-type diffuse gliomas, pediatric-type diffuse low-grade gliomas, pediatric-type diffuse high-grade gliomas, and circumscribed astrocytic gliomas. WHO grades are now expressed in Arabic numbers instead of Roman numerals. The 5th edition introduces 14 new gliomas and glioneuronal tumors and 8 other new tumors into the neuropathologic lexicon.36 The critical importance of identifying mutations other than the canonical IDH1 R132H mutation in diffuse gliomas, especially in patients younger than 55 years of age, is emphasized. Neuroradiologists must familiarize themselves with the updated WHO Classification of CNS neoplasms to function appropriately as part of the modern neuro-oncology clinical team.
Footnotes
Disclosure forms provided by the authors are available with the full text and PDF of this article at www.ajnr.org.
Indicates open access to non-subscribers at www.ajnr.org
References
- Received July 19, 2021.
- Accepted after revision November 8, 2021.
- © 2022 by American Journal of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Mesenchymal Nonmeningothelial Tumors of the CNS: Evolving Molecular Landscape and Implications for Neuroradiologists
- Ependymal Tumors: Overview of the Recent World Health Organization Histopathologic and Genetic Updates with an Imaging Characteristic
- High-Grade Astrocytoma with Piloid Features: A Dual Institutional Review of Imaging Findings of a Novel Entity
- Theranostics in Neurooncology: Heading Toward New Horizons
- Modeling Glioma Oncostreams In Vitro: Spatiotemporal Dynamics of their Formation, Stability, and Disassembly
- Comparison of the Diagnostic Performance from Patients Medical History and Imaging Findings between GPT-4 based ChatGPT and Radiologists in Challenging Neuroradiology Cases
- PiDeeL: Pathway-Informed Deep Learning Model for Survival Analysis and Pathological Classification of Gliomas
- Newly Recognized CNS Tumors in the 2021 World Health Organization Classification: Imaging Overview with Histopathologic and Genetic Correlation
- Amino Acid Tracer PET MRI in Glioma Management: What a Neuroradiologist Needs to Know
- Increasing Ciliary ARL13B Expression Drives Active and Inhibitor-Resistant SMO and GLI into Glioma Primary Cilia