
SUMMARY:
The nervous system is commonly involved in a wide range of genetic tumor-predisposition syndromes. The classification of genetic tumor syndromes has evolved during the past years; however, it has now become clear that these syndromes can be categorized into a relatively small number of major mechanisms, which form the basis of the new 5th edition of the World Health Organization book (beta online version) on genetic tumor syndromes. For the first time, the World Health Organization has also included a separate chapter on genetic tumor syndromes in the latest edition of all the multisystem tumor series, including the 5th edition of CNS tumors. Our understanding of these syndromes has evolved rapidly since the previous edition (4th edition, 2016) with recognition of 8 new syndromes, including the following: Elongator protein complex–medulloblastoma syndrome, BRCA1–associated protein 1 tumor-predisposition syndrome, DICER1 syndrome, familial paraganglioma syndrome, melanoma-astrocytoma syndrome, Carney complex, Fanconi anemia, and familial retinoblastoma. This review provides a description of these new CNS tumor syndromes with a focus on imaging and genetic characteristics.
ABBREVIATIONS:
- BAP1
- BRCA1–associated protein 1
- CNC
- Carney complex
- ELP1
- Elongator protein complex
- FA
- Fanconi anemia
- HNPG
- head and neck paraganglioma
- HNSCC
- head and neck squamous cell carcinoma
- MAS
- melanoma-astrocytoma syndrome
- MMNST
- malignant melanotic nerve sheath tumor
- NF
- neurofibromatosis
- NGS
- next generation sequencing
- PG
- paraganglioma
- PPB
- pleuropulmonary blastoma
- SDH
- succinate dehydrogenase
- SHH
- sonic hedgehog
- WHO
- World Health Organization
The World Health Organization (WHO) series of books (WHO Blue Books) and their Web site (https://tumourclassification.iarc.who.int) provide a routinely updated classification of multisystem tumors for standardizing diagnostic practices across the globe. This series is written by a wide group of multidisciplinary authors and editors, including radiologists who are experts in their respective fields, with the aim of providing the most updated nomenclature of tumors. During the past decade, there has been a rapid increase in our understanding of the variation in human genome sequences and identification of molecular information in tumor pathogenesis. This increase has led to better definitions of tumors that are difficult to classify on the basis of histomorphology and immunohistochemistry alone, and it also adds prognostic and therapeutic value. The diagnosis and management of tumors increasingly depend on their genetic causative mechanism, which ranges from simple sequence variants (mutations) to more profound genetic alterations such as translocations and gene fusions. As a result, the WHO, for the first time in 2022, released a dedicated book on genetic tumor syndromes, which forms the 14th and final volume of the 5th edition series on tumors. Besides the dedicated book, a new or updated chapter on genetic tumor syndromes has been added to the 5th edition of each organ system, including CNS tumors and head and neck tumors.1
The nervous system is frequently implicated in a wide range of hereditary tumor-predisposition syndromes with 10 syndromes described in the 4th edition (2016), including neurofibromatosis (NF), Von Hippel-Lindau disease, tuberous sclerosis, and Li-Fraumeni syndrome. Our understanding of these syndromes has evolved rapidly since the previous edition, with recognition of 8 new syndromes including the following: Elongator protein complex (ELP1)-medulloblastoma syndrome, BRCA1–associated protein 1 (BAP1) tumor-predisposition syndrome, DICER1 syndrome, familial paraganglioma syndrome, melanoma-astrocytoma syndrome (MAS), Carney complex (CNC), Fanconi anemia (FA), and familial retinoblastoma (Online Supplemental Data). The previous (4th) edition syndromes are preserved in the new (5th) edition except for Turcot syndrome, which has been removed, and its 2 subtypes (constitutional mismatch repair deficiency syndrome and familial adenomatous polyposis 1) are now identified as distinct entities. This change highlights the gradual shift from arbitrary and eponymic naming of the syndromes to a more gene-based system. Thus, a total of 19 CNS genetic tumor syndromes are now identified (Table), with the understanding that this is an evolving field and many changes may occur in the future.
There is more emphasis on the genetic pathogenesis of the previous and new entities in the 5th edition. Many of these syndromes are also described in other organ system books, with, however, greater emphasis on the nervous system in the book on CNS tumors.1,2 Also, many of the tumors can be seen in different tumor syndromes; for example, melanocytic nerve sheath tumors can be seen in MAS and CNC. Some of the newly recognized syndromes are well-known entities, (eg, FA); however, their involvement in CNS tumor predisposition has been better defined lately, necessitating inclusion in the chapter on CNS genetic tumor syndromes. Conventionally, the diagnosis of these syndromes relied on clinical features and family history; however, with the recent advancement in molecular genetics, the pathologist or geneticist might be the first to identify the syndrome. Radiologists are a vital part of the multidisciplinary team for these complex tumor syndromes and should be abreast of the changes in nomenclature. For example, it is important for the radiologist to know that most ELP1-related medulloblastomas are desmoplastic/nodular subtype and occur along the lateral cerebellar convexity with a low chance of recurrence postsurgery.3 These disorders are often highly complex, and patients are typically best served in specialized centers with broad multidisciplinary expertise.
In this article, we discuss the pathologic and imaging findings of the new tumor syndromes and highlight the inheritance pattern, because screening imaging examinations are increasingly being performed on the family members of these patients. Because genetic tumor syndromes are rare overall, the role of imaging-based artificial intelligence models in their prediction is still uncertain. However, some early studies have shown that there may be some benefit to the incorporation of radiomic data. Noortman et al,4 showed that PET-based radiomics moderately improved the distinction of cluster 1 and 2 paragangliomas (PGs) and from sporadic paragangliomas. Similarly, a recent meta-analysis noted that radiomics-based models showed robust performance in predicting medulloblastoma subgroups.5 Machine learning–based studies have previously shown promise in the identification of underlying genetic alterations in other tumors such as glioblastomas.6 The role of artificial intelligence–based studies in differentiating syndromic-versus-nonsyndromic tumors should be further explored in larger pooled cohorts.4,5
Genetics and Signaling Pathway
Alteration in the sequence of the human genome is the basis of heritable (constitutional) disorders further triggered by environmental factors. During the past few years, it has become clear that most genetic tumor syndromes are caused by a specific signaling pathway triggered by the genetic mutations and epigenetic factors. Figure 1 highlights the chromosomal distribution of genes involved in the old and new CNS genetic tumor syndromes, except for the succinate dehydrogenase (SDH) gene implicated in familial paraganglioma, which is located along the inner membrane of the mitochondria. The Table highlights the protein-signaling pathway for the new genetic syndromes. The growth factor receptor and the related signaling pathway are the major categories with the highest number of CNS and non-CNS tumor entities. These include NF, ELP1-related medulloblastoma syndrome, and nevoid basal cell carcinoma syndrome, among others. Some categories like the ubiquitin protein pathway have only the single entity of BAP1-related tumor predisposition syndrome. These changes in the signaling pathway and the altered protein product form the basis of molecularly targeted therapies and immunotherapy, an example being targeting the sonic hedgehog (SHH) signaling pathway through SHH inhibitors in medulloblastomas.7

This diagram highlights the chromosomal distribution of genes involved in the old (green boxes) and newly recognized (red boxes) CNS genetic tumor syndromes, with representation of 7 of the 8 new entities. The SDH gene implicated in familial paraganglioma is located along the inner membrane of mitochondria and is not depicted here. VHL indicates Von Hippel-Lindau syndrome.
The molecular and genetic information has facilitated the concept of “integrated diagnosis” to summarize all the information of a given tumor. This is a layered/tiered approach to the pathology report with a 4-line format, including integrated diagnosis, histologic diagnosis, WHO grade, and molecular information. For example, the integrated diagnosis for an ELP1-related medulloblastoma (Fig 2) would be reported as following: medulloblastoma desmoplastic/nodular morphology (histologic), WHO grade 4 (grade), with SHH activation (TP53 wild-type) and ELP1 germline mutation (molecular and genetic information). Molecular testing is a key component to the integrated diagnosis and overall characterization of many tumors, with a variety of techniques that can be used in a stepwise approach, including immunohistochemistry, fluorescence in situ hybridization, polymerase chain reaction, and next generation sequencing (NGS). Immunohistochemistry is a robust and economical test (<24 hours to complete) that is an excellent first-line method for determining the molecular subgroups. These are very efficient tests that detect the mutant protein (IDH, BRAF, H3) and serve as surrogate markers for genes encoding those proteins.

ELP1-related medulloblastomas in siblings. A 6-year-old boy (sibling A) with MR imaging revealing a heterogeneously T2 hyperintense (A, axial T2 TSE) mass with cystic changes and heterogeneous enhancement (B, coronal postcontrast). Histopathology with immunohistochemistry revealed a large-cell anaplastic-pattern WHO grade 4, SHH-activated (TP53 wild-type) medulloblastoma. The patient’s older brother, an 18-year-old boy (sibling B), was diagnosed with a similar tumor 7 years later. MR imaging revealed a right lateral cerebellar T2 hyperintense mass (C, axial T2 TSE) with solid contrast enhancement (D, coronal postcontrast). Pathology revealed an SHH-activated (TP53 wild-type) medulloblastoma with desmoplastic/nodular morphology. A neuro-oncology genetic panel (utilizing NGS technique) in both siblings showed biallelic inactivation of ELP1 due to somatic loss of chromosome arm 9q along with homozygous deletion of the tumor-suppressor gene PTCH1, confirming an ELP1 germline mutation. Both siblings remain disease-free (9 years postsurgery for sibling A, and 1 year for sibling B) (Online Supplemental Data).
Given all the genetic information now available, the WHO decided to rename the chapter from “Familial Tumor Syndrome” (4th edition) to “Genetic Tumor Predisposition Syndromes” (5th edition), stressing that many individuals might have the mutation and have a predisposition but may not develop these tumors. Many syndromes resulting in a predisposition to leukemia or lymphoma have been covered under “Hematolymphoid Neoplasia Genetic Predisposition Syndromes” and are not discussed in the chapter on CNS genetic tumor syndromes.2
Newly Recognized Genetic Tumor Syndromes
ELP1-Medulloblastoma Syndrome.
Medulloblastoma is the second most frequent malignant CNS tumor in children, and apart from sporadic occurrence, it can be associated with multiple cancer-predisposition syndromes like Turcot, Li-Fraumeni, and Gorlin syndromes. A mutation in a cancer-predisposition gene is estimated to be present in >40% of SHH-activated medulloblastomas, most commonly (14%) caused by biallelic alterations of ELP1.8 ELP1-medulloblastoma syndrome is an autosomal dominant disorder caused by pathogenic germline variants in the ELP1 gene (9q31.3). This is a tumor-suppressor gene that encodes for the protein ELP1, which is a part of the 6-subunit Elongator protein complex. This plays a key role in transcription of proteins that affect the structural framework (cytoskeleton) of the cell for a variety of cells including neurogenesis. This is a newly recognized genetic tumor syndrome in the WHO 5th edition of CNS tumors and is characterized by an increased risk of SHH-activated TP5 wild-type (grade 4) medulloblastomas during childhood. The median age at diagnosis is 6 years (range, 2–19 years), often with a family history positive for medulloblastomas.1,8 On histopathology, the desmoplastic/nodular pattern is most common (76%), followed by classic (18%) and large-cell/anaplastic (6%) morphology (Online Supplemental Data).3,8 The imaging features of the SHH subgroup are similar to those of the sporadic tumors and depend on the histologic subtype with the desmoplastic/nodular lesions located along the lateral cerebellar convexities and the less common, classic, or large-cell/anaplastic, variants located along the midline (Fig 2 and the Online Supplemental Data).
All the histologic subtypes are well-circumscribed and hyperintense on T2-weighted images. The enhancement is usually solid and intense, except the classic variant in which it can range from minimal and patchy to marked.9 ELP1 immunohistochemistry is a highly specific biomarker for identifying these medulloblastomas and is increasingly becoming a part of the neuropathologist’s panel to screen for tumor predisposition syndromes in children with medulloblastomas. Medulloblastomas have the potential for leptomeningeal spread, locally, or along the spinal axis and rarely extend outside the CNS. Leptomeningeal spread is usually along the cerebellar or cord surface, attached to the pia mater. Early studies indicate a favorable prognosis for ELPI-medulloblastomas with the 5-year overall survival rate of around 92%. Surveillance protocols have been established for many known syndromes like Gorlin syndromes and include annual MR imaging until 8 years of age; however, no formal protocol has yet been proposed for ELP1-medulloblastomas.8,10
BAP1 Tumor-Predisposition Syndrome
BAP1 tumor-predisposition syndrome is an autosomal dominant disorder caused by pathogenic germline variants in the BAP1 tumor-suppressor gene (3p21.1). This syndrome is characterized by a predisposition to tumors of multiple organ systems including the eye, pleura, peritoneum, kidneys, liver, and meninges. Uveal melanoma and mesothelioma are the most common tumors in this syndrome, seen in around 36% and 25% of patients, respectively.11 Other tumors in descending order of frequency include cutaneous melanoma, renal cell carcinoma, and basal cell carcinoma (Online Supplemental Data). Meningioma is the most common CNS tumor of this syndrome, seen in around 9% of patients (Fig 3).12 While most meningiomas are sporadic, many tumor-predisposition syndromes are increasingly being identified, including well-known conditions like NF-2 (or secondary to newly described entities like SMARCB1, SMARCE1 mutation, and BAP1 syndrome).13 Recent studies have shown that the patterns of mutations in many meningiomas are strongly associated with a distinct location and histologic subtypes; for example, anterior skull base meningiomas often have mutations in SMO, whereas posterior fossa and convexity meningiomas often have mutations in NF2.

Rhabdoid intracranial and spinal meningiomas with loss of BAP1 expression. Left lateral cerebellomedullary cistern enhancing mass (A, axial postcontrast T1, white arrow) in a 36-year-old man, extending into the hypoglossal canal, revealing atypical meningioma (CNS WHO grade 2) on histology, with rhabdoid features (B) and loss of BAP1 expression on immunohistochemistry (Online Supplemental Data). Coronal (C) and axial (D) contrast-enhanced MR images in a different patient depict an extramedullary cervical mass extending along the left neural foramen (arrows). Histopathology (not shown) revealed rhabdoid meningioma with loss of BAP1 expression.
Meningiomas with rhabdoid features represent a highly aggressive grade 3 malignancy with high rates of recurrence and mortality. Initial studies have shown that rhabdoid meningiomas often have BAP1 mutations, are clinically aggressive, and are more likely to recur, compared with BAP1-retained similar grade meningiomas.12,14 Recent advancements in the molecular-genetic taxonomy of meningiomas help in the identification of patients with inherited forms of meningiomas, with assessment of mutations, like SMARCB1 and BAP1, increasingly being performed at specialized centers, especially in cases of multiple meningiomas or younger age of onset. The BAP1 mutation can be easily assessed using simple and inexpensive immunohistochemical testing for BAP1 protein expression (Online Supplemental Data).1,13 Patients with BAP1 tumor-predisposition syndrome present with a younger age of onset compared with the sporadic counterpart for the same tumor type, with frequent occurrence of multiple cancers. For example, mesotheliomas with loss of BAP1 occur around 20 years earlier and are frequently peritoneal, rather than pleural, with a substantially different clinical course. The lifetime risk of developing cancer in this syndrome is as high as 80%–100%, with many patients developing multiple tumors.11,15 Most families with this germline mutation have at least 2 types of tumors in first- or second-degree relatives. Genetic counseling should be offered to patients with a history of ≥2 BAP1-related tumors or 1 tumor and a family history of similar tumors. Promising therapeutic avenues are being explored to specifically target BAP1-deficient tumor cells that are highly sensitive to novel therapies like inhibitors of enhancer of zeste homolog 2 (EZH2).16
DICER1 Syndrome
DICER1 syndrome is an autosomal dominant tumor predisposition syndrome caused by heterozygous germline loss-of-function mutation in the DICER1 gene (14q32.13), modifying the protein-coding genes by modulating microRNAs. This feature confers a lifetime risk of a variety of neoplastic and dysplastic lesions, classically associated with pleuropulmonary blastoma (PPB), a rare malignant tumor of the lung, seen primarily in children younger than of 6 years of age.1,17 Type I PPB is a purely cystic mass, occurring before 2 years of age. Type II is a solid-cystic tumor, while type III is purely solid; both types present from approximately 2 to 6 years of age and are malignant. Type III (solid) is the most aggressive with the highest mortality (up to 60%). Finally, “type I regressed” (Ir) is a cystic tumor lacking malignant cells and represents regressed type I PPB with no potential for malignancy.18 DICER1 syndrome occurs in children and young adults, with the most frequent phenotypes including PPB, multinodular goiter, cystic nephroma, and Sertoli-Leydig ovarian neoplasms (Online Supplemental Data).17
The most common CNS manifestation of DICER1 syndrome is brain parenchymal metastasis (from PPB) with the CNS being the most common site of distal metastasis. Surveillance MR imaging of the brain is recommended for up to 3 years after the diagnosis of aggressive forms of PPB (types II solid-cystic and III solid), because most of the brain metastases occur within 2 years of diagnosis. A few cases of spinal cord and leptomeningeal metastasis have also been reported.1,19 The primary CNS tumor manifestations of this syndrome include pineoblastoma, pituitary blastoma, embryonal tumor with multilayered rosettes, and DICER1-mutant primary intracranial sarcoma (Fig 4), which are indistinguishable on imaging and pathology from their sporadic counterparts. Although PPBs are the most common tumors in DICER1 syndrome, pituitary blastoma is most pathognomonic of this syndrome, with all reported cases to date arising in the setting of this syndrome.20

DICER1-mutant intracranial sarcoma. Sagittal T2 (A) and postcontrast (B) MR images in a 16-year-old adolescent girl reveal a dural-based left parasagittal T2-hyperintense enhancing mass, presumed to be a meningioma on imaging. Histopathology, however, revealed spindle and pleomorphic cells (C), and immunohistochemistry tests were negative for GFAP, OLIG2, SSTR2 (D), STAT6, S-100, and SOX10, arguing against glial, meningothelial, or melanocytic tumors. A Somatic Disease/Germline Comparator Exome sequencing panel revealed a pathogenic germline DICER1 mutation. Overall, the histomorphologic and immunophenotypic findings supported a sarcomatoid neoplasm. The additional presence of pathogenic DICER1 mutations was diagnostic of primary intracranial sarcoma, DICER1-mutant. Because the prognosis for patients with DICER1-mutant primary intracranial sarcoma remains unknown due to limited clinical data, a CNS WHO grade designation was not rendered, according to the 2021 WHO Classification of Tumors of the Central Nervous System.
Pineoblastoma is a rare primitive neuroectodermal grade 4 tumor, classically seen with RB1 in familial retinoblastoma, that has also been associated with the DICER1 mutation. Head and neck manifestations of this syndrome include nasal chondromesenchymal hamartoma, multinodular goiter, ciliary body medulloepithelioma, thyroblastomas, and thyroid carcinomas.1,2 Multinodular goiter occurring in patients younger than 18 years of age should prompt DICER1 testing, even in the absence of a family history or other syndromic tumors. Despite a higher risk of malignancy, most patients with the pathogenic germline DICER1 variants live healthy lives with tumors seen in <20% patients by the age of 50 years.17 Surveillance guidelines for these individuals include periodic chest radiographs and thyroid and pelvic sonography.21
CNC
CNC is a rare autosomal dominant syndrome with almost 100% penetrance, caused, in most cases (>70%), by heterogeneous inactivating pathogenic variants in the PRKAR1A gene (17q22-24). This regulates the cell signaling of the body, with protein kinase A (PKA) and subsequent loss of the tumor-suppressor function, resulting in overactivation of the cyclic adenosine monophosphate-protein kinase A (cAMP-PKA) pathway causing tumorigenesis.22 CNC was initially described in 1985, with myxomas, spotty pigmentation, and endocrine overactivity being the dominant features and should not be confused with the Carney triad. Myxomas may involve the heart (22%–53%), skin and mucosa (10%–30%), or bilateral breasts (25% of women with CNC). Nodular vertebral lesions on MR imaging may be seen in up to 32% of patients.23 These lesions are often multiple and can affect any part of the vertebral body. They show T1/T2 prolongation with postcontrast enhancement. Vertebral lesions are invariably nonaggressive on imaging and are presumed to represent osteochondromyxomas.23,24
The main nervous system manifestation is malignant melanotic nerve sheath tumor (MMNST), seen in 8%–10% of adults with the syndrome, often along the paraspinal sympathetic chain and the gastrointestinal tract, less commonly intracranial and dermal. Historically considered benign, these were reclassified as malignant tumors in the 2020 WHO Classification of Tumors as well as in the 2021 WHO Classification Tumors of the Central Nervous System. Psammomatous MMNST (which contains psammoma bodies) accounts for about one-half of all MMNSTs, and approximately 50% of these are associated with CNC. A recent series noted a median age of 34.5 years (range, 25–54 years) at presentation.22,25 Lesions may show calcifications on CT and demonstrate T1/T2 shortening on MR imaging due to the presence of melanin (Fig 5 and Online Supplemental Data).26 CNC is also characterized by endocrinal neoplasms, most frequently being primary pigmented nodular adrenocortical disease, resulting in Cushing syndrome, seen in 26%–58% of patients with CNC. Somatotroph pituitary neuroendocrine tumor (adenoma) may occur in 10%–18% of adults. These tumors often secrete growth hormone and are generally diagnosed early, often at a microadenoma stage.22⇓⇓-25

MMNST in a 26-year-old woman with CNC. Coronal MR images (A and B) reveal a mass along the left S1–S2 neural foramen with intrinsic T1-hyperintense content (A, T1-weighted, non-fat-saturated, arrow) with peripheral enhancement and a central nonenhancing component (B, postcontrast with fat saturation, arrow). Note marked expansion of the neural foramen and erosive osseous changes along the inferior edge on coronal CT (C, arrow). Histopathology reveals a spindle cell (D, white arrow) neoplasm with psammomatous bodies (D, black arrow). Immunohistochemistry was positive for SOX10, S-100, and MelanA, confirming the diagnosis of MMNST. Neoplastic cells showed complete loss of PRKAR1A, confirming an association with CNC (Online Supplemental Data).
MAS
MAS is a rare autosomal dominant tumor-predisposition syndrome caused by a germline mutation of the CDKN2A tumor-suppressor gene (9p21.3), which codes for cell-cycle regulators and the apoptosis pathway (P16INK4A and P14ARF). MAS is characterized by an increased risk of multiple tumors, including cutaneous melanoma, astrocytoma, nerve sheath tumors, pancreatic cancer, and squamous cell carcinoma of the oropharynx.27 In 1993, Kaufman et al28 identified familial MAS in a 3-generation family with members having melanomas or astrocytomas. Subsequent studies have found more families with similar syndromes, linking an increased risk of dysplastic nevi, cutaneous melanoma, and nervous system tumors, especially astrocytomas. MAS is considered a glioma-predisposition syndrome with diverse histologic and genetic features. Dysplastic nevi and melanomas associated with MAS are predominantly cutaneous, rather than mucosal, acral, or uveal. Astrocytomas associated with MAS include pleomorphic xanthoastrocytoma (Online Supplemental Data) and diffuse astrocytic gliomas, ranging from low-grade diffuse astrocytomas to high-grade (glioblastoma), which are typically located in the cerebral hemispheres or cerebellum.27 Patients can present with numerous glial neoplasms, with reported cases of simultaneous development of pleomorphic xanthoastrocytoma, diffuse astrocytoma, and paraspinal nerve sheath tumors.29 These tumors cannot be differentiated from their sporadic counterparts on the basis of histopathology, and detection of the CDKN2A mutation is required for diagnosis of the syndrome. Some cases with large deletions involving CDKN2A and CKDN2B have shown manifestations like neurofibromas, giant cell tumors of bone, and multiple primary cancers resembling NF1 or Li-Fraumeni syndrome. Clinical outcomes and standardized surveillance protocols for MAS-associated tumors are still being investigated. Regular dermatologic examinations, brain imaging, and genetic counseling are usually recommended for monitoring various cancers and guiding patients and their families.27⇓⇓-30
Familial Paraganglioma Syndrome
Familial paraganglioma syndrome represents a group of inherited cancer syndromes characterized by the development of PGs and pheochromocytomas. These are highly vascular neuroendocrine tumors arising from the extra-adrenal neural crest tissue or the adrenal medulla, respectively. Head and neck PGs (HNPGs) develop within the parasympathetic chain, most commonly along the ninth and tenth cranial nerves (jugular foramen, carotid body), followed by the middle ear cavity. Nearly 95% of these are nonsecretory, contrary to secretory PGs, which mainly involve the adrenal medulla (pheochromocytomas). About 30%–40% of PGs in adults are hereditary, characterized by a younger age at presentation and a higher risk of multiplicity and metastatic spread, along with a higher incidence of extra-adrenal tumors.31 Apart from the genetic mutations in Von Hippel-Lindau disease, multiple endocrine neoplasia type 2, and NF-1, nearly 10 separate genes have been implicated in this syndrome. Of these, the mitochondrial succinate dehydrogenase (SDH) complex subunit genes (SDH A-D) and 1 complex cofactor, SDHAF2, are collectively responsible for nearly one-half of all germline mutations. Inactivating mutations in any of the SDH genes result in the accumulation of succinate and the formation of reactive oxygen species (pseudohypoxia cluster). HNPGs are largely either sporadic or associated with germline SDH variants. Of these, carriers of SDHD/SDHAF2 mutations have the highest risk of multifocal HNPGs.32,33
Overall, contrast-enhanced MR imaging has a sensitivity and specificity of nearly 95% and 99%, respectively, but CT has a better sensitivity to identify tumors of <1 cm and those in the jugulotympanic region.34 MR imaging perfusion may help differentiate HNPGs from non-PG tumors such as schwannomas, with the former showing markedly higher vascularity and perfusion. SDH-intact (no mutation) tumors show higher mean and normalized ADC values (normalized mean ADC, 1.73 versus 1.31; P < .001) compared with SDH-mutated tumors.35 Gallium 68DOTATATE PET demonstrates high lesion detectability for SDH-mutated PGs and is generally considered first-line imaging for HNPGs, especially when suspecting metastatic or multifocal disease (Fig 6) or identifying peptide-receptor therapy candidates. This has emerged as the new standard of care for functional imaging of neuroendocrine tumors. It is highly sensitive for the detection of both solitary and metastatic PGs, substantially more so than 123I metaiodobenzylguanidine scintigraphy, CT, or MR imaging, particularly for demonstrating metastatic bone disease.34,36

HNPGs in 2 different patients, one with SDH mutation (patient A) and the other with normal SDH expression (patient B). Patient A has familial paraganglioma syndrome with multiple HNPGs and a left adrenal phaeochromocytoma (A, arrowheads) on a 68Ga-DOTATATE PET scan, with complete loss of SDH expression on immunohistochemistry (C). A solitary HNPG along the right carotid artery is noted on a 68Ga-DOTATATE PET scan in patient B (B, arrow) with normal SDH expression on pathology (D).
FA
FA is a clinically heterogeneous multisystem disorder with an incidence of around 1 per 100,000 live births and is more common in Ashkenazi Jews, Afrikaners, and Spanish gypsies. The underlying etiology is the inability of the affected cells to repair DNA interstrand crosslinks.37 At least 22 genes have been implicated (FANCA-FANCW) with most inherited in an autosomal recessive pattern and a smaller minority being X-linked recessive (2%) or autosomal dominant (RAD51).38 This syndrome is characterized by developmental abnormalities in the major organs, inherited bone marrow failure characterized by pancytopenia, and a high predisposition to cancer.1,37 The median age at diagnosis is about 7 years, though earlier diagnosis is becoming common with higher disease awareness and prenatal screening. The most common neoplasms associated with this syndrome are myeloproliferative conditions, including myeloid leukemia, squamous cell carcinoma of the head and neck (HNSCC), Wilms tumor, and medulloblastoma. Leukemia, specifically acute myeloid leukemia, is the most common cancer in patients with FA. Overall, HNSCC is the most common solid tumor in patients with FA, with the incidence being 500–700 times higher than in the general population. Patients with FA generally develop HNSCC at a younger age (20–50 years), usually with a more aggressive clinical course and advanced disease at presentation. HNSCC is the leading cause of death in patients with FA in adulthood. Thus, surveillance for HNSCC in FA is recommended starting at a young age and continuing through life.39
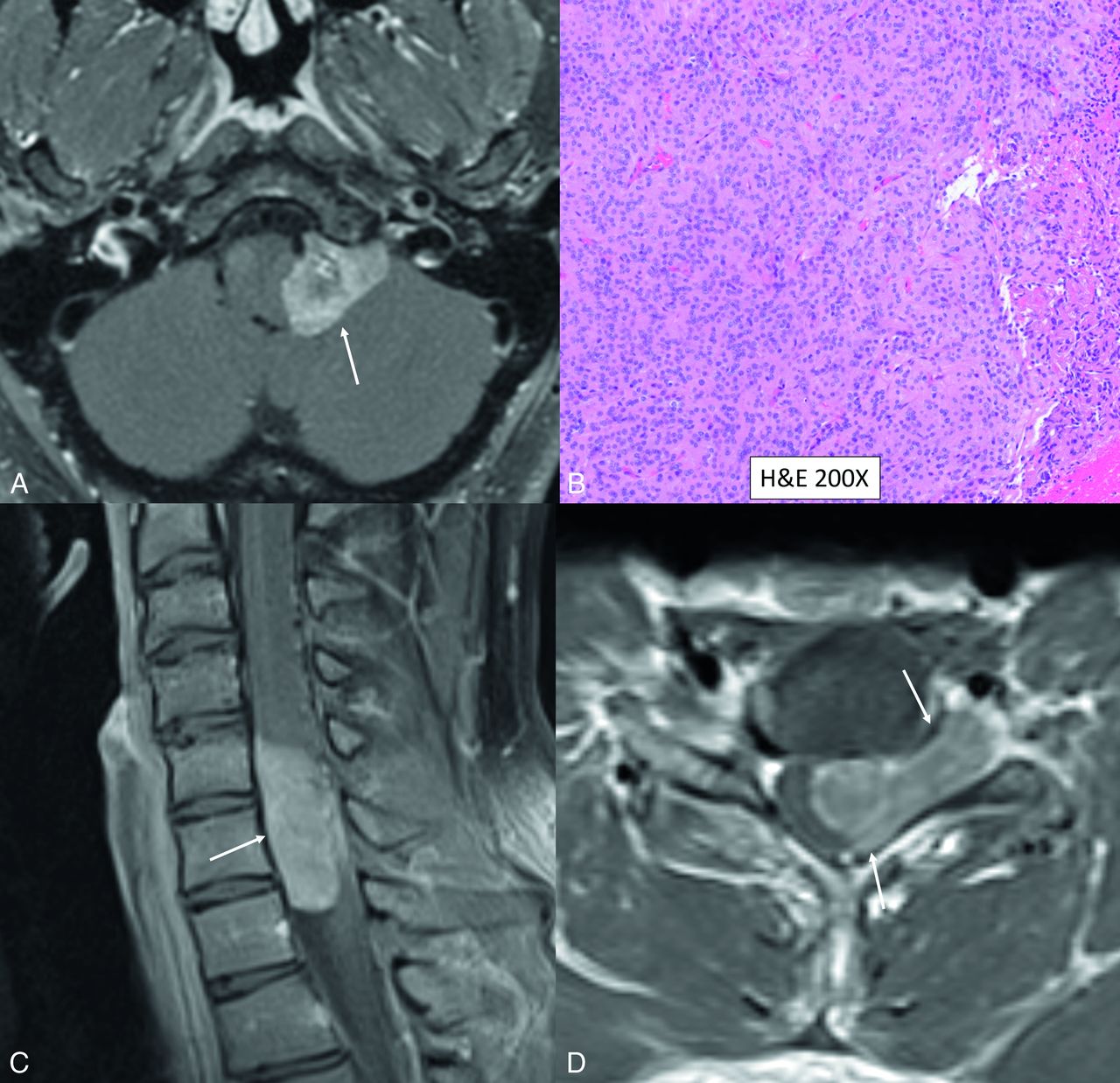
Medulloblastoma is the characteristic CNS tumor in FA secondary to biallelic germline mutations in BRCA2 or PALB2.1,3,40 A few cases of cerebroretinal vasculopathy and leukoencephalopathy have been reported in literature. These are characterized by recurrent space-occupying brain lesions that can mimic glial neoplasms along with retinal vasculopathy, with patients presenting in their early twenties (Fig 7 and Online Supplemental Data).41 Rare cases of embryonal tumors and glioblastomas have also been reported with these mutations. These mutations are also associated with cancers of the breast, ovaries, and pancreas. Medulloblastomas arising in the setting of FA are usually the large-cell/anaplastic histologic type, with SHH-activated TP53-mutant being the most common molecular subtype. These medulloblastomas with germline BRCA2 or PALB2 mutations have a poor prognosis and a significantly higher risk of the patient developing metastases after treatment.40,42 The poor prognosis is due to the aggressive biology of the tumor and because these patients cannot tolerate high-dose chemotherapy, given the underlying myelodysplastic condition. The essential diagnostic criterion for FA is positive findings on the diepoxybutane test, wherein chromosomal breakage is seen after exposure to diepoxybutane in leukocytes. Identification of pathogenic germline mutations in a FANC gene through NGS is, however, more sensitive and desirable.1,38

Cerebroretinal vasculopathy and leukoencephalopathy in a 20-year-old man with FA. MR imaging including a sagittal T2-weighted image (A), axial diffusion (B), T2 TSE (C), and postcontrast (D) images reveal a large area of white matter edema with heterogeneous enhancement and central necrosis in the right frontal lobe. The primary radiographic differential included a high-grade glial neoplasm, and a biopsy was performed. Histopathology revealed severe vasculopathy exhibiting vascular hyaline changes and perivascular and transmural chronic inflammation. The background white matter demonstrated extensive vacuolization and gliosis with necrosis surrounding the vessels. No evidence of JC virus or Epstein-Barr virus was identified by in situ hybridization, and no fungal organisms were detected. Follow-up MR imaging (Online Supplemental Data) after 6 months of immunosuppressive treatment showed marked reduction in the right frontal edema and enhancement, however, with new areas of edema and enhancement in the left parieto-occipital lobe. Advanced veno-oclusive retinopathy with neovascularization was noted on fundoscopy (Online Supplemental Data).
Familial Retinoblastoma
Retinoblastoma is the most common intraocular malignancy in children with small, blue, round-cell morphology and a classic histologic pattern characterized by hyperchromatic small, round, blue cells arranged in sheets, nests, and trabeculae (rosettes). This CNS tumor can be secondary to a germline RB1 mutation (heritable) or somatic mutation (RB1 sporadic, MYCN-driven). MYCN oncogene–related retinoblastomas are the least common, are nonhereditary, and are almost always unilateral, with a very early age of presentation.43 Familial retinoblastoma is secondary to a germline RB1 mutation with bilateral ocular disease in 60% of patients along with synchronous or metachronous intracranial tumors (pineal or suprasellar). Histologic features of pineal and ocular tumors are identical in familial retinoblastoma. The combination of intraocular retinoblastoma (unilateral or bilateral) and a histologically similar-but-independent focus of brain tumor (pineal region or suprasellar) is called “trilateral retinoblastoma.” Children with familial retinoblastoma have a particularly high incidence of and earlier onset of trilateral retinoblastoma.44,45 Most of these tumors are seen in children younger than 3 years of age, with familial tumors arising at an earlier age (younger than 1 year) compared with the sporadic counterpart.
These tumors arise from the inner or outer retinal layers, and can be endophytic or exophytic, causing vitreous invasion or retinal detachment, respectively. The most important predictive factors for recurrence include the extent of retrolaminar optic nerve involvement and choroidal invasion (Online Supplemental Data).1 Patients with familial retinoblastoma, whether treated by radiation or not, are at higher risk of subsequently developing sarcomas as a second neoplasm. With better understanding of tumor genetics, a clear association of the RB1 gene in oncogenesis of osteosarcomas has been established.29,46 Patients with familial retinoblastomas should be screened for osteosarcomas (10–20 years of age) and soft-tissue sarcomas (10–50 years).47
CONCLUSIONS
Multiple new genetic tumor syndromes have been described in the new 5th edition of the WHO Classification of Tumors of the Central Nervous System, highlighting the rapid advances in our understanding of the molecular and genetic tumor markers, including the diversity of clinically relevant molecular types and subtypes. Recognition of these genetic tumor-predisposition syndromes is of great importance to patients and their families. Surveillance protocols for individuals affected by these syndromes are being established, with consensus from international experts. The radiologist plays a vital role in identification, surveillance, and follow-up of CNS tumors seen in these genetic syndromes and for screening examinations for family members. Radiologists should be aware of the tumors associated with these syndromes, the inheritance pattern, and the latest guidelines for screening of family members and can expect to see higher numbers of screening examinations in their practice.
| WHO Classification, 4th Edition 2016 | WHO Classification, 5th Edition 2022 | ||
|---|---|---|---|
| Familial Tumor Syndromes (Chapter 16) | Genetic Tumor Syndromes Involving the CNS (Chapter 14) | ||
| New Syndromes | Genetic Pathway | Most Common Nervous System Tumors | |
| NF type 1 | ELP1-medulloblastoma | Growth factor receptor | Medulloblastoma |
| NF type 2 | BAP1 tumor predisposition | Ubiquitin protein | Meningioma |
| Schwannomatosis | DICER1 syndrome | microRNA regulation | Metastasis (from pulmonary blastoma) |
| Von Hippel-Lindau syndrome | CNC | PKA signaling pathway | Malignant melanotic |
| Nerve sheath tumor | |||
| Tuberous sclerosis | Melanoma-astrocytoma | Cell cycle and apoptosis | Glioma |
| Li-Fraumeni syndrome | Familial paraganglioma | Oxidative stress (Krebs) | Paraganglioma |
| Cowden syndrome | FA | DNA repair and genomic stability | Medulloblastoma |
| Turcot syndrome | Familial retinoblastoma | Cell cycle and apoptosis | Retinoblastoma |
| Constitutional mismatch repair deficiency syndrome | |||
| Familial adenomatous polyposis 1 | All entities from 4th edition kept in the new edition, except Turcot syndrome | ||
| Nevoid basal cell carcinoma syndrome | Constitutional mismatch repair deficiency (previously subtype under Turcot syndrome) | ||
| Rhabdoid tumor syndrome | Familial adenomatous polyposis 1 (previously subtype under Turcot syndrome) | ||
Newly recognized CNS genetic tumor syndromes in the 5th WHO classification with updates on genetic pathway
Footnotes
Disclosure forms provided by the authors are available with the full text and PDF of this article at www.ajnr.org.
Indicates open access to non-subscribers at www.ajnr.org
References
- Received July 11, 2023.
- Accepted after revision September 14, 2023.
- © 2024 by American Journal of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.