
In fiscal year 2001, the National Institutes of Health (NIH) total research expenditures for stroke reached nearly $239 million. A significant focus of stroke research is the development of therapeutic strategies that prevent neuronal death and improve recovery, but, unfortunately, few therapeutic strategies have successfully moved from basic science laboratories to the clinic. In this review, we discuss reasons why neuroprotective therapies that work in the laboratory fail in clinical trials. By analyzing research methods and difficulties extrapolating from stroke models to clinical disease, we identify ways to improve therapeutic development. In addition, we review some of the promising neuroprotective strategies in development.
Methods of Preclinical Research
The rationale seems simple: identify pathophysiologic processes in disease models and target them therapeutically. In practice, however, this paradigm is difficult to accomplish in a clinically relevant fashion. Basic scientists study many pathologic processes in in vitro systems and animal models. Cell culture models are useful because they provide well-controlled systems in which experimental variables can be manipulated easily and conclusions are clear. When cells are placed in culture, however, changes in cell structure, receptor populations, and gene expression occur that alter cellular responses from their normal in vivo state. In vitro systems may be most useful for studying fundamental cell biology that is less likely to be altered by culture systems. In many cases, it is unclear how in vitro systems compare with human disease.
Animal models have advantages over in vitro systems because the observed cells are in their natural state. Unfortunately, drawing conclusions from animal systems is challenging because it is difficult to manipulate just one variable. To keep the number of variables to a minimum, animal models of human disease are designed to be reproducible among subjects. By simplifying disease models, the experimental disease becomes fundamentally different from the human condition. Models are designed so specifically that they may work differently among breeds of the same species (1).
Animal Models of Cerebrovascular Disease
Global Ischemia
Global ischemia models reduce blood to the entire brain, mimicking cerebral ischemia from cardiac arrest or severe hypotension. In rats, global ischemia may be produced by two-vessel or four-vessel occlusion or cardiac arrest. The two-vessel occlusion model involves tying ligatures around the rat’s carotid arteries and decreasing mean arterial pressure to 50 mm Hg by controlled exsanguination. Blood is immediately reinfused after 10 minutes and the ligatures removed. Four-vessel occlusion involves permanent occlusion of vertebral arteries with transient occlusion of the carotid arteries. The cardiac arrest model attempts to induce ischemia in a more clinically relevant fashion through the pharmacologic induction of cardiac arrest for 7–10 minutes, followed by injection of epinephrine and chest compression. These injuries result in neuronal necrosis in selective brain regions including the cerebral cortex, hippocampus, striatum, and cerebellum. Cell death is complete 3–7 days after injury, and because of this delay, therapies can be tested for their ability to prevent cell death (2).
Focal Ischemia
Most focal ischemia models reduce blood supply to focal brain areas by large-artery occlusion. Techniques include middle cerebral artery (MCA) suture ligation, intraluminal occlusion, or photothrombosis. Suture ligation involves simply tying off the MCA after surgical exposure (3). Intraluminal MCA occlusion is performed by inserting a stiffened suture into the external carotid artery, through the internal carotid artery and up to the MCA base (4). Photothrombotic MCA occlusion is produced by injecting rats with a photosensitive dye such as Rose Bengal or Erythrosin B and focusing an argon laser onto the exposed MCA. The laser-dye interactions produce singlet oxygen, damaging the vessel wall and forming a platelet thrombus (5). Suture models can be studied with permanent or transient occlusion by removing the suture. The photothrombotic model also has been studied in permanent and transient models by using a UV laser to dethrombose the occluded vessel (6). The photothrombotic model was developed as a more clinically relevant model of disease, since the occlusive lesion is a natural platelet clot.
Embolic Stroke
Most models of cerebrovascular disease study pure ischemia. Photothrombotic MCA occlusion adds the complexity of a thrombotic process that more closely mimics human disease. To take the photothrombotic model one step further, the laser-dye system was modified to produce nonocclusive common carotid artery (CCA) thrombosis. A nonocclusive platelet thrombus develops when the laser is focused on the CCA. Small platelet emboli break off of the main thrombus, travel downstream, and occlude distal cerebral vessels. Infarcts from this model are generally smaller than pure ischemic models, and because the mechanism of injury is embolic, the insult can vary among animals. Although CCA thrombosis is somewhat variable, it has been used to study cerebrovascular consequences of embolic stroke (7) and the effects of multiple emboli over time (8), as well as to test neuroprotective agents (9). An alternative thromboembolic model involves taking blood from the animal and permitting it to coagulate ex vivo. Clots are then injected into the carotid circulation.
Clinical Trials
Once a therapeutic strategy has amassed considerable evidence from the preclinical literature, it may be attempted in clinical trials. The NIH divides clinical trials into four phases. Phase I trials test a drug on a small number (ie, 20–80) of usually healthy people to determine overall safety, safe dosages, and side effects. Phase II trials are performed with larger numbers (ie, 100–300) of patients who have the condition the drug is designed to treat; phase II trials evaluate safety and efficacy. Phase III trials involve large numbers of patients (ie, 1000–3000+) and evaluate side effects and efficacy alone or in comparison to those of other treatments. Phase IV trials occur after U.S. Food and Drug Administration approval and further evaluate adverse events, efficacy, and optimal use. Kidwell et al (10) reviewed 178 clinical trials for ischemic stroke from 1990 to 1999, more than half of which studied neuroprotective agents. They found that less than 2% of trials met strict criteria defining them as having a positive outcome, though 23% suggested the agent was beneficial. Disappointing clinical trial results are not limited to the stroke literature. According to Faden (11), the past 50 years of clinical trials in traumatic brain injury resulted in no convincing evidence of beneficial neuroprotective agents. With these realizations, investigators began a great deal of introspection into fundamental methods of research to assess reasons for discrepancy between preclinical and clinical outcomes (12, 13).
Preclinical Pitfalls
The following pitfalls are summarized in Table 1.
Why neuroprotective agents work in animal models and not in patients
Anesthesia and Neuroprotection
Almost all animal protocols require anesthesia in any situation that causes pain. Although necessary for humanitarian reasons, volatile anesthetics such as isoflurane and halothane reduced infarct volumes early after focal ishemia and selective neuronal necrosis at 14 days (14, 15). Animal studies using both anesthesia and neuroprotective agents are actually studying the combination of two therapies. These complications are important to consider when interpreting results.
Dosing
Drug doses used in clinical trials often are based on animal studies. Determining the minimum effective dose should entail generating dose-response curves in multiple stroke models by different groups of investigators. Generating dose-response curves is frequently overlooked because doing so quickly increases the cost in terms of time, personnel, materials, and number of animals; however, it is critical to determine both the minimum effective dose and any hazards of using higher or lower doses. Small-animal studies rarely yield information on harmful side effects because small animals tolerate drugs very well, are not routinely examined for systemic evidence of side effects, and are frequently sacrificed early after treatment, so long-term consequences are unknown. To address these issues, organizations such as the Stroke Therapy Academic Industry Roundtable recommended generating dose-response curves in models of both small and large animals (16). The more species tested, the more likely investigators are to detect harmful side effects. In addition, larger animals might be more prone to similar side effects as humans.
Difficulties in dosing and side effects are particularly marked in studies with the glutamate receptor antagonists selfotel (CGS 19755) and MK-801. Doses of selfotel around 40 mg/kg were determined to be neuroprotective in a variety of animals, including gerbil (17) and rabbit (18) models of ischemia; however, phase IIa trials found the limit of safe doses in patients with stroke to be 1.5 mg/kg (19). Higher doses led to psychotomimetic effects characteristic of glutamate receptor antagonists. Trials with the agent continued, and two phase III trials were eventually suspended owing to an early increase in mortality in the selfotel-treated group and a trend toward increased 90-day mortality (20). Dawson et al (21) retrospectively studied side effects of glutamate receptor antagonists, including selfotel, in rats with permanent MCA occlusion. They used a vestibulomotor test (rotorod) to evaluate adverse neurologic effects. They determined that agents such as selfotel have detectable side effects that give them therapeutic ratios of less than 1, whereas more promising neuroprotective therapies have ratios greater than 1 (21). Careful detection of side effects and the generation of dose-response curves may prevent drugs that are bound for failure from going to clinical trials. Modifying neuroprotective strategies based on therapeutic ratios may improve clinical trial success rate.
A potential new development in designing clinical trials was summarized by Lees (22). Adaptive randomization design is a new technique based on Bayesian statistics that will allow trial designers to use fewer patients in determining minimal effective doses. Although these techniques are not appropriate for conclusions regarding efficacy, they will reduce the numbers of inadequately treated or overtreated patients (22).
Therapeutic Window
Time between injury and start of therapy is often discrepant between preclinical and clinical studies. Therapeutic manipulations generally work best when administered before and immediately after the insult. Experiments with animal models often begin with a pretreatment protocol. If the therapy works, it is tested at different intervals from injury onset. Most effective therapies work best within 15–30 minutes; rarely are they effective more than 3 hours after injury (23). Although there is little reason to assume the therapeutic window in small animals relates to the window in humans, less than 3 hours seems to fit both paradigms. Unfortunately, it is difficult to get patients to the hospital, evaluated, and enrolled in a clinical trial within 3 hours of symptom onset. More than 17,324 patients were screened to enroll the 624 subjects in the National Institute of Neurological Disorders and Stroke (NINDS) recombinant tissue-type plasminogen activator (t-PA) trial, and most were excluded because of the time window (13, 24). In the European Cooperative Acute Stroke Study II, which looked at recombinant t-PA therapy with a treatment window between 0 and 6 hours, more than 80% of patients were treated between 3 and 6 hours and no effect was detected (25). Had the NINDS trial chosen a longer window, a difference might not have been detected and recombinant t-PA might not have made it to clinical use. To avoid erroneously discarding a beneficial therapy, future trials should either strictly enforce enrollment or stratify patients into groups based on time-to-treat. Conducting randomized, double-blind, placebo-controlled studies that stratify patients based on time-to-treat will require larger numbers of subjects and take longer to complete; however, these studies may give investigators the best chance of detecting efficacious therapies.
Preclinical studies often begin with a pretreatment design to improve chances of detecting an effect. Clinical correlates to pretreatment studies include administering neuroprotective agents to long-term high-risk patients or during procedures that carry risk of stroke such as carotid endarterectomy or those involving the aortic arch. Most procedures have relatively small risks of complications, so these studies would likely take a long time to acquire sufficient numbers of subjects but may help establish the neuroprotective potential of new therapeutics.
Therapeutic Targets
A fundamental question in translating results of animal experiments to clinical use is whether or not the pathophysiologic processes in animal models correlate with those in human disease. Differences may arise for the following reasons: differences in anatomy and physiology, differences in pathophysiologic response to injury, or differences between injury mechanisms in animal models and those in human disease. The first two problems have been addressed by using multiple species in each model. As a therapy advances toward clinical trials, larger animals, including primates, may be used to better predict response in humans. The third problem can be addressed by testing therapeutics in multiple models. Therapies that work in many models are more likely to target fundamental pathomechanisms and apply to human disease. Unfortunately, investigators often gain expertise in only a few similar models with questionable applicability to human disease. Furthermore, having multiple, independent laboratories replicate studies provides a stronger rationale for moving a therapy to the clinic as the probability of having multiple false-positive studies are reduced.
Evidence from Animal Models: The Ischemic Penumbra
The ischemic penumbra can be broadly defined as that region of the ischemic zone that is potentially salvageable (26). As such, it is the focus of current research into neuroprotective strategies for stroke treatment. The time until the area becomes irreversibly damaged is the therapeutic window. The penumbra resides around the core infarct and is characterized by hemodynamic, metabolic, and molecular alterations (27, 28). Most of the biochemical and histologic descriptions of the penumbra were studied in models of MCA occlusion in which single large-vessel occlusion permits collaterals to feed areas adjacent to the ischemic focus, producing a large penumbra amenable to neuroprotective agents.
Embolic models such as CCA thrombosis produce thrombi in the CCA that embolize and occlude both large and small distal vessels. It is unclear whether or not a significant penumbra exists in these models. Instead of occluding one large vessel and permitting collaterals to feed the “penumbral” region, collateral flow may be reduced by embolic occlusion of collateral vessels. In another rat model of embolic stroke, injected thrombotic clots were observed occluding large vessels at early time points and microvessels at later time points (29). The authors concluded that the initial clots in large vessels break up and embolize distally to smaller vessels.
Other subtle pathophysiologic features of embolic models have been described. For instance, vasoactive factors produced from thrombotic processes occurring in damaged vessels may reduce flow to significant portions of the cerebral hemisphere, further limiting collateral flow and the penumbral territory (30). In addition, platelet embolization results in alterations of vascular reactivity through regulation of endothelial nitric oxide synthase, suggesting that vessels exposed to platelet emboli may have altered hemodynamic control (7).
Preconditioning studies with ischemic stroke or CCA thrombosis also have illustrated differences between models. After a brief episode of either focal or global ischemia that causes no histologic damage (the preconditioning stimulus), molecular and cellular events occur that protect the brain from future insults in a phenomenon known as ischemic preconditioning. Thus, when a damaging insult (usually global ischemia) is induced after a preconditioning stimulus, there is less damage than if global ischemia was induced without preconditioning. CCA thrombosis causes very small focal infarcts and was studied to determine if it could act as a preconditioning stimulus. In contrast to preconditioning with focal or global ischemia, CCA thrombosis made the brain more vulnerable to damage after a second insult. When a damaging episode of global ischemia was induced after CCA thrombosis, the ischemic lesion was much more severe than global ischemia alone, even at sites distant from embolic infarcts (31). These results suggest that platelet embolization makes the brain more susceptible to future insults, even in locations distant from embolic infarcts. This example illustrates how differences in stroke models result in very different phenomena and is likely analogous to what is occurring in patients with different types of stroke.
Clinical Studies of the Penumbra
Many studies report evidence of penumbral regions in patients after stroke (32–36) and have begun to address the following key questions: How can the penumbra be identified? What proportion of patients with stroke have a viable penumbra? How large is the ischemic penumbra? How long does the penumbra last? Will saving the penumbra improve outcome? Answers to these basic questions will enable clinicians and investigators to identify patients most likely to benefit from neuroprotective therapies.
Radiologic studies have demonstrated regions representing ischemic penumbra in some stroke patients. To define a region as the penumbra, hemodynamic and metabolic parameters associated with impending infarction were identified in both animal and human studies. Positron emission tomography (PET) helped identify tissue with reduced cerebral blood flow (CBF) and regional cerebral metabolic rate of oxygen that represented infarcted and penumbral regions (34, 35). Baron (36) combined PET, repeated CT imaging, and clinical correlations to further characterize the penumbra in man. The author concluded that penumbral tissue is variably represented in different patients. Penumbra was detected up to 16 hours in some and was absent by 5 hours in other patients.
Heiss et al (37) combined PET with 60-mCi H215O and 20-mCi [11C]flumazenil to measure both CBF and benzodiazepine (BDZ) receptor density, respectively. BDZ receptor density is used as a marker of neuronal integrity. They established 95% probability limits of infarction (4.8 ± 0.53 mL/100 g/min) and survival (14.1 ± 1.83 mL/100 g/min) for CBF and for [11C]flumazenil binding. They concluded that measuring these parameters identifies tissue compartments amenable to neuroprotective therapies. In their cohort of 10 patients, 13% of the final infarct volume had sufficient CBF and neuronal integrity to be capable of rescue (37). In three patients, this area was over 45%, suggesting that some patients are more likely to benefit from neuroprotective therapies than others. A complication in this study was that patients were assessed an average of 6 hours after onset of symptoms, but one would expect the percentage of salvageable tissue to decrease over time, particularly after the first 3 hours (33).
White versus Gray Matter
Preclinical studies have largely studied neuroprotection of gray matter since rats have a higher proportion of gray to white matter than humans (13). Neuroprotective therapies that work well on gray matter do not necessarily have the same effect on white matter, which makes up a large portion of human stroke. For example, MK-801 was neuroprotective against selective neuronal necrosis in photothrombotic focal stroke (38) and in global ischemia (39), but did not attenuate white matter damage in a model of focal ischemia in cats (40). The α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) antagonist SPD 502, however, has demonstrated efficacy in reducing both white and gray matter damage in rat focal ischemia (41). Neuroprotective effects against white and gray matter should be evaluated before progressing to clinical trials. MR imaging techniques that distinguish gray and white matter injuries may be useful to stratify patients for trials that are beneficial against their particular lesion.
Sex-Specific Injury Response
Experimental studies frequently use only male animals to reduce variability between mixing sexes and avoid having to account for the estrus cycle in experimental design. Recent data have established sex differences after experimentally induced stroke (42). Studies with models of embolic stroke global and focal ischemia describe reduced injury in intact female versus male rodents (43–45). These data also suggest that ovariectomized female rodents have injuries more consistent with those of male rodents (45, 46). Testosterone was studied by comparing castrated versus intact male rats and was reported to worsen outcome after focal ischemia (47). In contrast to these data, the Women’s Health Initiative study recently reported that estrogen and progesterone hormone replacement therapy increases risk of stroke (48). The estrogen-only arm of the study is awaiting completion. Because stroke patients are likely to be postmenopausal, it is unclear whether or not sex differences due to estrogen introduce variability into clinical trials. Recent experimental data in a model of traumatic brain injury reported that therapeutic hypothermia does not improve outcome in females as it does in males (49). It is unclear whether these results are similar in stroke models. Thus, sex differences might complicate neuroprotection trials with regard to both outcome and therapeutic response.
Problems with Clinical Trials
The following sections are also summarized in Table 1.
Patient Selection
In contrast to preclinical studies that use young, healthy animals under controlled physiologic conditions, patients have various causes of stroke and have comorbidities. When therapies targeting ischemic penumbra are given to patients who lack a penumbra, the study is no longer sensitive to finding a difference between treated and untreated patients. Radiologic techniques that can screen for patients with salvageable areas are being incorporated into clinical trial selection criteria. Validation of these radiologic screening systems is already under way. The combination of lesion volume on diffusion-weighted images, NIH Stroke Scale score, and time from symptom onset to imaging was recently validated as a predictor of patient outcome (50). The Alberta Stroke Program Early CT Score divides the MCA territory into 10 sections, and 1 point is subtracted for each section containing hypoattenuation or evidence of early ischemic changes. Thus, a normal score is 10 and lesion of the entire MCA territory is 0. A score of less than 7 yielded a 14-fold increase in risk of symptomatic intracerebral hemorrhage in patients treated with the thrombolytic ateplase, compared with scores above 7 (51).
Identifying penumbra is the key toward developing useful stratification guidelines. In a retrospective study, Schlaug et al (32) were able to identify penumbra in 25 patients presenting within 24 hours of hemispheric stroke onset, and they operationally defined the penumbra by measuring regional CBF (rCBF) and regional cerebral blood volume (rCBV) with diffusion- and perfusion-weighted MR imaging. To determine parameters best able to identify penumbral regions at greatest risk of infarction, Schaefer et al (52) examined diffusion- and perfusion-weighted images in 30 patients with stroke, between 1 and 12 hours after symptom onset. They found that rCBF ratios (lesion-to-contralateral) are most useful when compared with parameters such as rCBV, mean transit time, apparent diffusion coefficient, signal intensity on diffusion-weighted images, or fractional anisotropy. Regional CBF distinguishes between penumbra that infarcts and hypoperfused tissue that recovers. The combined efforts of these and other investigators are establishing the utility of neuroradiologic techniques to improve clinical trials and enhance stroke treatment.
Outcome Measures
Choosing similar outcome measures would aide comparisons between preclinical and clinical studies (13). Preclinical studies often evaluate early injury response, whereas late outcomes are most clinically important. Experiments that find reduced infarct size early after injury fail to establish whether treatment attenuates or simply delays injury. Preclinical studies often use infarct volume to determine efficacy, whereas clinical studies rely on behavioral measures. Behavioral measures were developed to evaluate functional recovery in animals. Rats often regain much of their behavioral functioning over time, even without treatment, and a battery of tests are most effective at detecting persistent deficits. After CCA thrombosis for example, rats have sensorimotor (forelimb placing) deficits up to 3 days after injury, no difference in vestibulomotor (beam balance) skills, and only subtle impairments in cognitive tasks (Morris water maze) at 48 hours but not 21 days after injury (53). After MCA occlusion, sensorimotor deficits recovered by day 30 but cognitive deficits remained up to 5 weeks (54). Recommendations to avoid these pitfalls include ensuring that all neuroprotective agents have established short- and long-term histopathologic and behavioral benefits (16).
Selecting outcome measures have been equally problematic in clinical trials. Duncan et al (55) conducted a review of outcome measures in 51 large neuroprotection trials. Only 29 trials had defined outcome measures and time frames for their end point. There was considerable variability among studies, including 15 different impairment measures, 11 activity measures, one quality-of-life-health status measure, and eight miscellaneous measures designed by the investigators. Well-defined and standardized outcome measures would aid in meta-analysis and cross comparisons between trials.
Objective radiologic data are beginning to be used as outcome measures. Although physicians agree that functional outcomes are most important, reduced lesion volumes lead to functional improvement. Radiologic outcomes could help steer research in appropriate directions by detecting subtle therapeutic benefits. Functional improvements may require substantial tissue salvage best accomplished by multiple therapies, each making small contributions to limit infarction. Therapies that yield a clear radiologic benefit but no functional improvement should not be discarded. Reevaluating doses and time of administration may improve efficacy. Flaws in trial design and patient selection may have contributed to failures in detecting improved functional outcome. Warach (56) argued that clinical trials of 100–200 patients with use of radiologic outcomes might have sufficient power to evaluate efficacy, whereas 5–10 times as many patients are required in phase III trials. Thus, studies with radiologic outcomes may be a cost-effective approach for deciding whether or not to plan a phase III trial.
Neuroprotective Therapies
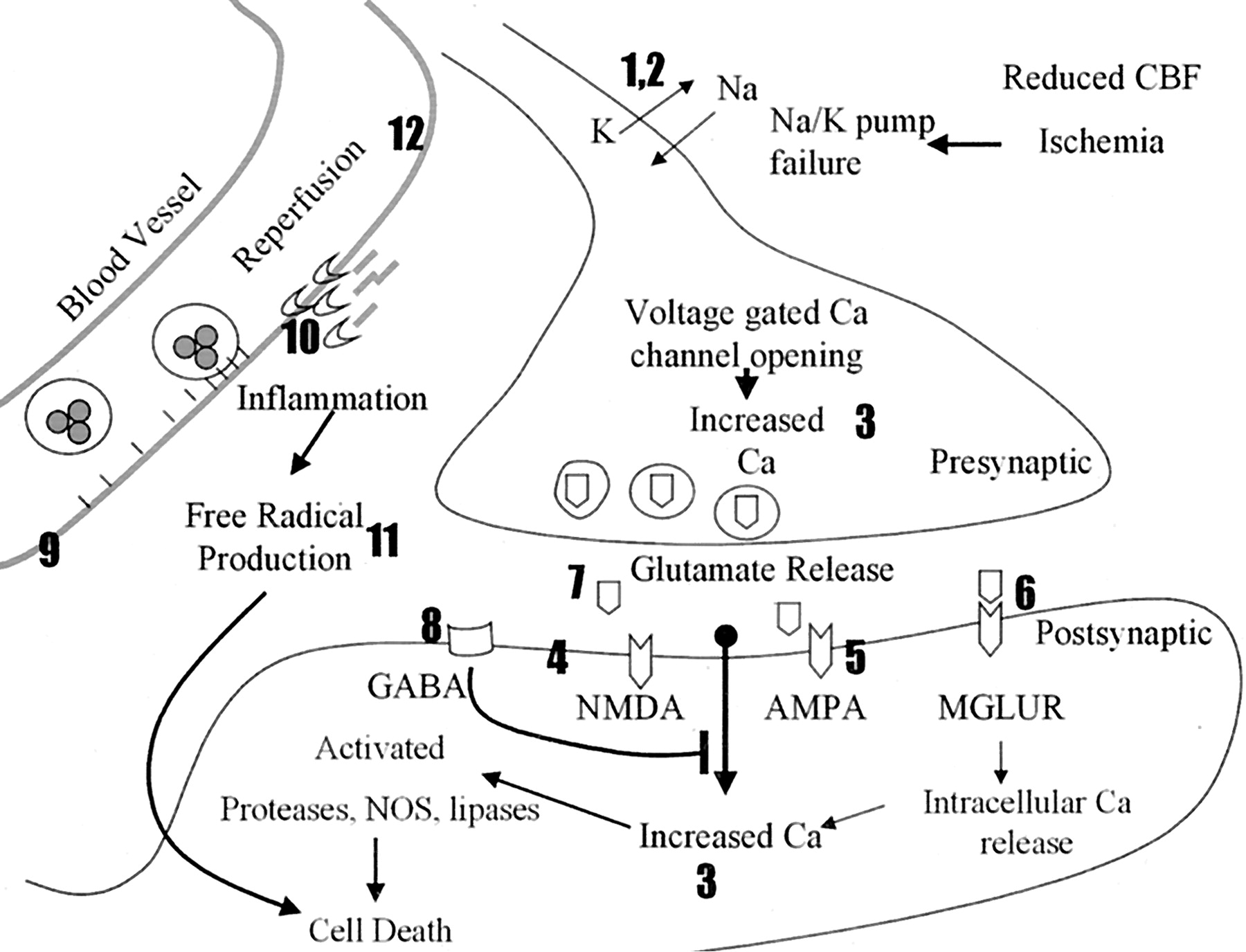
Neuroprotective strategies can be divided into those that limit the ischemic cascade or those that reduce reperfusion injury (Fig 1 and Table 2). Neurons die under ischemic conditions when delivery of oxygen and an energy source (ie, glucose) are not sufficient to meet their metabolic demands. Therefore, ischemic neurons must either receive increased blood flow or their metabolic demands need to decrease. The most important step in limiting ischemia is the quick restoration of blood flow that occurs either naturally or with the aid of thrombolytics such as t-PA. Before complete restoration of blood flow, therapies such as hypothermia may decrease metabolic demand (57) and extend neuron survival (58). Once depleted of energy, membrane-bound ion pumps fail, leading to membrane depolarization, sodium and calcium entry, and neurotransmitter release (59). Dendritic glutamate release activates downstream neurons, leading to calcium entry, cellular damage, apoptosis, and further release of toxic molecules. Agents that modulate ion channels and glutamate receptors target this pathway (60, 61). Reperfusion injury was observed by comparing infarct volumes in animal models of transient and permanent ischemia (6, 62). Reperfusion, while critical to limit ischemia, contributes to cell death by enhancing production of free radicals, inflammation, and blood-brain barrier breakdown; each has been targeted by therapies (60, 61, 63, 64).

Schematic of the ischemic cascade. The numbers 1–12 correspond to the numbers 1–12 at each class of therapy in Table 2. When the presynaptic neuron becomes ischemic, it depolarizes, opening sodium (Na) and potassium (K) channels (1, 2). This leads to opening of calcium (Ca) channels and influx of calcium (3). The presynaptic cell releases glutamate that activates NMDA, AMPA, and MGLURs (4–6), which permits calcium entry in the postsynaptic cell. NAALADase inhibitors limit the amount of glutamate available for release (7). GABA agonists inhibit depolarization and prevent calcium entry (8). During reperfusion, upregulated adhesion molecules attract inflammatory cells that contribute to vessel occlusion and cytotoxin production (9). MMPs degrade the basement membrane around vessels, enhancing inflammation and edema (10). Inflammatory cells, ischemic neurons, and glia produce free radicals that contribute to cell damage and death (11). HMG-CoA reductase inhibitors (statins) stabilize the endothelium through upregulation of endothelial nitric oxide synthase (NOS, 12).
Outcomes in experimental and clinical trials of various neuroprotective agents
Limiting the Ischemic Cascade
Sodium Channel Blockers.—
Blocking sodium channels prevents damage from both initial ion channel failure and excitotoxic activation (59). Experimental studies with phenytoin, carbamazepine, lamotrigine, sipatrigine (619C89), and riluzole showed benefits in a variety of stroke models (65–68). Sipatrigine demonstrated efficacy in pretreatment but not posttreatment studies (68). Clinical trials with sipatrigine and fosphenytoin were discontinued without success because of toxicity and no efficacy, respectively (68–70).
Potassium Channel Modulators.—
Drugs such as BMS-204352 open potassium channels thereby hyperpolarizing cells, protecting them from excitotoxicity. BMS-204352 was developed by Bristol Myers Squibb (Princeton, NJ) and decreased infarct volume in a rat model of permanent focal ischemia (69), but showed no efficacy in phase III trials (71, 72).
Calcium Channel Blockers.—
Calcium enters into damaged neurons through voltage-gated calcium channels and N-methyl-D-asprate (NMDA)-sensitive ligand-gated channels, and release from intracellular stores. Cytoplasmic calcium activates enzymes and second messenger cascades that contribute to cell death. Activated proteolytic enzymes break down elements of the cytoskeleton, leading to protein aggregation. Calcium-mediated lipolysis damages membranes and along with nitric oxide synthase activation provides nitric oxide and fatty acid substrates for free radical production. Apoptotic cascades are stimulated by the rise in calcium through mitochondrial permeability. Glutamate release is stimulated by calcium-dependent exocytosis and has toxic effects on neighboring cells. For these reasons, calcium channel blockers have been studied as potential neuroprotectants (73).
Experimental studies have been conducted with a variety of calcium channel blockers in a number of stroke models. (S)-emopamil is a phenylalkylamine with additional serotonin receptor 5-HT2 activity. Pretreatment with (S)-emopamil improved neuronal survival by about 2-fold after global ischemia in rats, but was not effective when administered 30 minutes after treatment (74). In permanent focal ischemia models, (S)-emopamil decreased infarct volume when administered up to 1 hour after onset of ischemia (75).
Nimodipine is a calcium channel blocker used clinically for treating hypertension that has been studied as a neuroprotectant in animal models (76) and clinical trials (73). It decreased infarct volume by 50% in a rat model focal ischemia (76). Experimetal trials were mixed, however, as no benefit was observed after 20 minutes of four-vessel occlusion global ischemia (77). At least 14 clinical trials of nimodipine in ischemic stroke were conducted since 1984. Nine trials found no effect, one trial found short-term worsened outcome with treatment, and four trials found positive outcomes. Positive outcomes included improved survival in men (78), memory (79), clinical course (80) and Mathew score among certain subgroups (81). Two clinical trials with flunarizine found no statistically significant improvement in outcome (82, 83). A meta-analysis of 29 trials totaling 7665 patients treated with calcium channel blockers found no benefit for ischemic stroke (73). Despite this discouraging analysis, the calcium channel blocker dantrolene has been recently discussed as a potentially beneficial agent (60). Dantrolene prevents glutamate cytotoxicity in neuron cultures by reducing the intracellular calcium concentration (84). In animal models, it reduced cortical necrosis by 85–95% after ischemic stroke in rats (85), protected against delayed neuronal death in gerbils (86), but had no effect after 11 minutes of complete ischemia in dogs (87).
Glutamate-Mediated Excitotoxicity
Under ischemic conditions, excessive glutamate is released and binds to receptors on adjacent cells. Glutamate receptors may be metabotropic or inotropic, acting through G-protein or ligand-gated ion channels, respectively. Once activated, calcium influx results in activation of proteolytic enzymes and apoptosis, and induces action potentials leading to more glutamate release. Glutamate receptors can be inhibited in a competitive (competes with glutamate for the same receptor site) or noncompetitive (antagonist binds elsewhere on the receptor) fashion to limit the excitotoxic cycle.
NMDA Antagonists.—
The NMDA receptor is an inotropic, calcium-conducting receptor attributed to much of the excitotoxic response following ischemia (88). A number of antagonists have been studied in experimental and clinical trials (60, 61, 63, 89). Although animal studies were very promising, most clinical trials were stopped because of phencyclidine-like effects such as hallucinations and agitation. The noncompetative antagonist MK-801 (dizocilpine) improved outcome in models of focal ischemia producing either no effect or between 13–78% reductions in infarct volume, depending on experimental conditions (90, 91). Suzuki et al (92) reported significantly reduced infarct volume in young rats, with no efficacy in aged rats; this finding suggested that age-related changes may alter drug efficacy. In models of global ischemia, significant increases in CA1 neuron survival and behavioral improvement were also observed, especially when MK-801 was combined with hypothermia (39, 93); however, beneficial findings have not been observed consistently in all models. No benefit was observed in a rat model of photothrombotic MCA occlusion (94) or after transient focal ischemia in primates (95). Both MK-801 and dextromorphan, another noncompetative NMDA inhibitor, found protective effects in experimental studies, but clinical trials were terminated early because of phencyclidine-like side effects and lack of efficacy (61, 96).
Aptiganel (Cerestat, CNS1102) is also a noncompetative NMDA antagonist that decreases infarct volume and behavioral deficits up to 120 minutes after focal ischemia (97). Safety and tolerability studies in patients found neuroprotective plasma levels in healthy volunteers and patients with ischemic stroke (98). Unfortunately, a phase III clinical trial of aptiganel treatment within 6 hours of stroke was terminated early because of no efficacy and a trend toward higher mortality in treated patients.
CP101,606 (Ceresine) is a noncompetative NMDA antagonist selective for the NR2B subunit. CP101,606 improved behavioral outcome and decreased infarction volume 20–30% 2 hours after thromboembolic stroke in rats (99). A 63% reduction in infarct volume was observed in a feline model of focal ischemia with pretreatment and continuous infusion (100). Initial trials found that CP101,606 was well tolerated in patients with traumatic brain injury and intracerebral hemorrhage (61, 101).
Other NMDA antagonists with evidence of efficacy in animal models include selfotel (CGS 19755), eliprodil, and NPS-1506. Selfotel is a competative NMDA antagonist that was discontinued after lack of efficacy in two phase III trials (102). Eliprodil is a competative NMDA antagonist that also blocks calcium channels. It reduced neurologic deficit and infarct volume by about 50% in a rat model of thrombotic stroke. The combination of eliprodil and t-PA reduced deficit and infarct volume by 70% and 89%, respectively; however, a phase III trial was terminated owing to lack of efficacy (61). NPS-1506 is a noncompetative NMDA receptor antagonist that inhibits active channels in a voltage-dependent fashion. It was found to be neuroprotective in animal models of hemorrhagic stroke, brain trauma, and transient and permanent focal ischemia with 33–57% decreases in infarct volume at effective doses. Phase I trials were completed and found no phencyclidine-like side effects at neuroprotective doses (103).
AMPA Antagonists.—
Non-NMDA antagonists are being studied for their potential role as neuroprotectants. YM-872 (zonampanel) administered up to 2 hours after permanent and transient focal ischemia decreased infarct volume and neurologic deficit by 30–40% at 1 week (104, 105). YM-872 also improved outcome in a rat model of thromboembolic stroke and enhanced t-PA efficacy (106). Overall, animal models have reported a 20–60% decrease in infarct volumes (60). YM-872 is being studied in two large trials, the AMPA Receptor Antagonist Treatment in Ischemic Stroke Trial (ARTIST+) and AMPA Receptor Antagonist Treatment in Ischemic Stroke with MR imaging (ARTIST MR imaging). The ARTIST+ trial compares t-PA with and without YM-872 treatment. The ARTIST MR imaging trial is measuring outcome at 28 days by using T2-weighted MR imaging with a fluid-attenuated inversion-recovery, or FLAIR, sequence and at 90 days by clinical criteria.
N-acetylaspartylglutamate (NAAG) Inhibitors.—
NAAG is a stored form of glutamate, released by the enzyme N-acetylated-á-linked acidic dipetidase (NAALADase). Inhibitors of NAALADase include 2-(phosphonomethyl)pentanedioic acid, or 2-PMPA, and a more lipophilic analog GPI5232. They decrease glutamate release and are neuroprotective in neuron culture as well as in animal models of transient focal ischemia (decreased infarct volume around 50%) (107–110).
Metabotropic Glutamate Receptor (MGLUR) Modulators.—
There are eight subtypes of MGLUR, divided into three groups. The group I MGLURs activate the phosphoinositide/Ca++ cascade, while groups II and III depress intracellular systems such as adenylate cyclase and calcium entry. At least nine group I antagonists, six group II agonists, and three group III agonists demonstrated neuroprotective properties. BAY 36–7620 is a recently developed group I antagonist with oral bioavailability and good blood-brain barrier penetration (111). It was neuroprotective after subdural hematoma in rats, but statistical significance was not reached after focal ischemia (112).
Magnesium.—
Magnesium may be protective through a variety of mechanisms, including inhibition of noncompetitive NMDA receptors, neurotransmitter release, voltage-gated calcium channels, and cortical spreading depression, and it promotes vasodilatation (60). It is particularly appealing because of its low cost, widespread availability, and relative safety. Magnesium was protective in models of thromboembolic, transient, and permanent focal ischemia, with decreases in infarct volume of 25–61%. At least six clinical trials showed promise for magnesium therapy (113). Three large clinical trials are under way to further evaluate magnesium’s efficacy. The Intravenous Magnesium Efficacy in Stroke trial is recruiting 2700 patients to assess death and disability when magnesium is administered within 12 hours of stroke onset. The Magnetic Resonance in Intravenous Magnesium Efficacy in Stroke trial will determine the ability of magnesium to reduce infarct volume determined with MR imaging. The Field Administration of Stroke Therapy—Magnesium Phase III trial will examine if magnesium is effective when administered by ambulance personnel between 15 minutes and 2 hours after symptom onset.
γ-Aminobutyric Acid (GABA) Agonists.—
GABA is an inhibitory neurotransmitter, generally thought to work in opposition to glutamate. Thus, GABA may be neuroprotective by counteracting glutamate-mediated excitotoxicity. Numerous studies found neuroprotective effects in global and focal ischemia (114). Clomethiazole was the most recent candidate for GABA therapy. The phase II Clomethiazole Acute Stroke Study found no benefit, but the drug was well tolerated. The phase III trial called the Clomethiazole Acute Stroke Study in ischemic, hemorrhagic, and t-PA-treated patients was discontinued after demonstrating no efficacy (115).
Vascular-Targeted Therapeutics
Carotid stenosis, platelet embolization, and components of the thrombotic cascade damage the cerebral vasculature and contribute to the pathophysiology of acute stroke. Drugs targeting the endothelium such as serotonin antagonists and adenosine agonists were recently reviewed (116). Although these agents were promising in experimental animal models, their clinical utility remains uncertain.
Free Radical Scavengers.—
Free radicals are molecules with one or more unpaired electrons that participate in oxidation-reduction reactions. They are produced during ischemia-reperfusion, diffuse into neighboring tissue, and damage blood vessels, neurons, and glia. Vascular damage from these molecules may promote blood-brain barrier permeability, endothelial dysfunction, and inflammation. Free radicals react with nitric oxide produced by nitric oxide synthase isoenzymes to form peroxynitrite, a highly reactive molecule. More detailed descriptions of free radical biology after ischemia and the role of nitric oxide in these reactions are reviewed elsewhere (117, 118).
Free radical scavengers may be beneficial for protecting both the vasculature and the brain parenchyma, but some agents target one better than the other. For example, tirilazad was more effective at reducing blood-brain barrier damage, whereas U-101033E was more effective at preventing neuronal cell death and infarction in models of global and focal ischemia although both agents are members of the lazaroid class (119). Although most agents are tested in the early phase, there are some experimental data on their use at longer intervals after injury. U-101033E significantly reduced infarction size at 4 hours but not at 24 hours after permanent focal ischemia (119). NXY-059 reduced infarct size when administered 3 hours after transient focal ischemia. Treatment at 6 hours but not at 3 hours improved behavioral deficits at 1 day but not at 2 days after injury. The scavenger á-phenyl-tert-butyl-nitrone successfully reduced blood-brain barrier permeability and infarct size 12 hours after focal ischemia (120). Although most agents were tested in rodent models, NXY- 059 has successfully improved the behavioral and histopathologic consequences of permanent MCA occlusion in marmosets (121), raising hopes that these treatments will be clinically applicable.
Clinical trials with free radical scavengers have had limited success after acute ischemic stroke, but have had more success in the treatment of subarachnoid hemorrhage. Patients administered nicaraven 5 days after subarachnoid hemorrhage had a reduced rate of delayed ischemic neurologic deficits. A number of trials examined tirilazad after stroke, and a consensus group determined that patients were likely worse off in the treated groups. Numerous side effects may have complicated any benefit of the drug (122). NXY-059 was well tolerated in patients with stroke and is currently being studied for efficacy by AstraZeneca Pharmaceuticals (Wilmington, DE).
Statins.—
Statins (3-hydroxy-methylglutaryl coenzyme A, or HMG-CoA, reductase inhibitors) inhibit the rate-limiting enzyme of cholesterol synthesis in the liver and are successfully used to treat hypercholesterolemia. Meta-analysis of multiple clinical trials reported a 16–30% reduction in stroke (123) that was independent of serum cholesterol (124). Guidelines from the National Stroke Association recommend the use of statins in some patients (125), and a pharmacoeconomic analysis of statin treatment suggests that it is a cost-effective means for preventing cardiovascular disease and stroke (126). Hepatic and skeletal muscle toxicities are the most common complications. Statins are reported to prevent platelet aggregation, restore endothelium-dependent dilatation, reduce oxidative damage, and inhibit matrix metalloproteinase (MMP) degradation of the extracellular matrix (127). Mevastatin upregulates endothelial nitric oxide synthase messenger-RNA and protein in mice, which promotes vasodilatation and decreases platelet aggregation. Treated mice also had reductions in infarct size and behavioral deficits after transient MCA occlusion (128). Thus, statins may become an important part of cholesterol-independent, preventive stroke therapy.
Antiinflammatory Therapy
Hematogenous inflammatory cells contribute to stroke pathology through two basic mechanisms. They form aggregates that reocclude vessels after reperfusion or enter infarcted tissue and exacerbate cell death through production of cytotoxins (129). To function in either of these mechanisms, cells must first attach to endothelial cells in cerebral vessels. Cell adhesion molecules such as selectins, integrins, and intercellular adhesion molecules (ICAMs) permit endothelial-inflammatory cell interactions. Increased adhesion molecule expression was reported after experimental stroke in rat models of thromboembolic stroke, as well as permanent and transient focal ischemia in rats (130) and baboons (131). Some studies in stroke patients report increases in selectins, ICAMs, and vascular cell adhesion molecules after stroke, although these results are not consistent (129). Contributing to both mechanisms is the production of various cytokines such as interleukin-1, tumor necrosis factor á, and others that stimulate adhesion molecule expression and activation of inflammatory cells.
Anti-Adhesion Molecule Therapy.—
Pre- and posttreatment with antiselectin antibodies successfully decreased infarct volume up to 70% after 2 hours of transient focal ischemia and improved CBF (132, 133). The anti-ICAM-1 antibody 1A29 decreased infarct areas after transient but not permanent focal ischemia. Cotreatment with the granulocyte-depleting antibody RB6-8C5 further improved outcome (134). Recently, a clinical trial of the murine anti-ICAM-1 antibody enlimomab worsened neurologic score and mortality in patients (135). A follow-up study using the murine anti-rat ICAM-1 antibody in rats also found an increase in infarct volume and no efficacy. It is believed that immune activation in response to the foreign mouse protein probably accounted for the clinical and follow-up experimental results (136).
Matrix Metalloproteinases.—
MMPs are enzymes that break down components of the extracellular matrix. Although their implication in angiogenesis suggests a potential benefit, they may enhance blood-brain barrier breakdown after stroke, promote hemorrhage, and increase inflammation. MMP inhibitors such as batimistat (BB-94) and KB-R7785 decreased infarct volume in mice after permanent focal ischemia (137). BB-1101 reduced edema 24 hours after focal ischemia in rats and reduced hemorrhage in both rats and rabbits (138–140). MMP inhibitors have been evaluated in patients for their antiangiogenic properties and are well tolerated (141).
Therapeutic Hypothermia
The effects of small variations in brain temperature have been tested in a number of stroke and brain injury models. For example, intraischemic hypothermia after transient global ischemia protected the CA1 hippocampus and dorsolateral striatum from neuronal necrosis (142) and attenuated cognitive and sensory motor deficits (143). In dogs, mild hypothermia at 34°C also resulted in significant improvement in neurologic function after cardiac arrest (144). Mild temperature reductions dramatically reduced infarct volume after transient focal ischemia (145, 146), whereas profound temperature reductions (24°C) or extended periods of mild hypothermia were required to reduce infarct volume after permanent focal ischemia (147, 148).
One of the limitations of postischemic hypothermia appears to be the therapeutic window. Although dramatic protection is observed if hypothermia is induced during or immediately after the ischemic insult, lesser degrees of protection are observed as a delay in the initiation of hypothermia is demonstrated. On the basis of current experimental data, a 2–3-hour hypothermic window has been consistently reported in the literature (149). This indicates that cooling the patient needs to begin soon after the primary insult.
Several clinical studies have been initiated to test the beneficial effects of therapeutic hypothermia in various patient populations (150–153). Recently, two multicenter trials were published that reported benefits of therapeutic hypothermia (154, 155). In the European study, nine centers in five countries participated with a total enrollment of 275 patients. Systemic hypothermia for 24 hours combined with passive rewarming resulted in a favorable neurologic outcome in 55% of hypothermic patients compared with 39% of normothermic patients. The fact that two independent studies resulted in basically the same positive findings and conclusions is extremely positive in the field of therapeutic hypothermia.
Recently, feasibility studies have been initiated to determine whether systemic hypothermia can be produced in stroke patients (153, 156). Schwab and colleagues (153) induced moderate hypothermia in 25 patients with severe ischemic stroke of the MCA territory. Patients were kept at 33°C for 48–72 hours by surface cooling. In that pilot study, hypothermia was shown to reduce intracranial pressure. During the rewarming period, however, brain hemorrhage and secondary intracranial pressure were documented. Investigators are currently studying the rewarming phase to determine how to control intracranial pressure and cerebral perfusion pressure during the critical posthypothermic period (157). Also, relevant to the preclinical data, studies have shown reduced CSF levels of glutamate in stroke patients who were cooled compared with the levels in normothermic subjects (158).
An important area of investigation is the development of better methods of patient cooling. In the past, bags of ice and body blankets have been used to produce a surface cooling with some success. As the necessity for more critical control of a patient’s temperature becomes apparent, other strategies must be developed to accomplish this goal. To this end, new strategies for surface cooling, as well as endovascular cooling catheters, are being developed and tested (159). These cooling devices are demonstrating that periods of hyperthermia (fever) in patients after central nervous system injury can be reduced in frequency; also these devices are being used to produce systemic hypothermia. This is especially important since an experimental study demonstrated that a small increase in brain temperature worsens outcome (160). The continued development of this important field will improve the way we administer hypothermia and, hopefully, provide better protection and treatment for patients with neurologic disorders. Combination therapies including hypothermia, t-PA, antiinflammatory, and antiapoptotic agents, as well as caffeine or alcohol, may represent important treatment directions.
Summary
Neuroprotection is a worthwhile pursuit given the tremendous costs posed on society by neurologic disease. Numerous difficulties in translating experimental data to clinical use have plagued the field, as demonstrated by the list of therapies discarded from clinical trials. Physicians and scientists from around the world have reevaluated past problems and are looking forward to improvements in clinical investigations. Combining thrombolytics with neuroprotective strategies, including antiexcitotoxic agents, free radical scavengers, vascular protective agents, antiinflammatories, and hypothermia, shows great promise and a bright future for treating cerebrovascular disease.
Footnotes
Supported by National Institutes of Health grant NS27127-11.
References
- Received July 31, 2003.
- Accepted after revision August 29, 2003.
- Copyright © American Society of Neuroradiology





{kind=link}