
Abstract
BACKGROUND AND PURPOSE: Zhu-Tokita-Takenouchi-Kim syndrome is a severe multisystem malformation disorder characterized by developmental delay and a diverse array of congenital abnormalities. However, these currently identified phenotypic components provide limited guidance in diagnostic situations, due to both the nonspecificity and variability of these features. Here we report a case series of 7 individuals with a molecular diagnosis of Zhu-Tokita-Takenouchi-Kim syndrome, 5 ascertained by their presentation with the neuronal migration disorder, periventricular nodular heterotopia.
MATERIALS AND METHODS: Individuals with a molecular diagnosis of Zhu-Tokita-Takenouchi-Kim syndrome were recruited from 2 sources, a high-throughput sequencing study of individuals with periventricular nodular heterotopia or from clinical diagnostic sequencing studies. We analyzed available brain MR images of recruited individuals to characterize periventricular nodular heterotopia distribution and to identify the presence of any additional brain abnormalities.
RESULTS: Pathogenic variants in SON, causative of Zhu-Tokita-Takenouchi-Kim syndrome, were identified in 7 individuals. Brain MR images from these individuals were re-analyzed. A characteristic set of imaging anomalies in addition to periventricular nodular heterotopia was identified, including the elongation of the pituitary stalk, cerebellar enlargement with an abnormally shaped posterior fossa, rounding of the caudate nuclei, hippocampal malformations, and cortical anomalies including polymicrogyria or dysgyria.
CONCLUSIONS: The recurrent neuroradiologic changes identified here represent an opportunity to guide diagnostic formulation of Zhu-Tokita-Takenouchi-Kim syndrome on the basis of brain MR imaging evaluation.
ABBREVIATIONS:
- PVNH
- periventricular nodular heterotopia
- VUS
- variant of uncertain significance
- WES
- whole-exome sequencing
- WGS
- whole-genome sequencing
- ZTTK syndrome
- Zhu-Tokita-Takenouchi-Kim syndrome
Understanding of the distinctive phenotypic features of a rare syndromic disorder facilitates earlier diagnosis. This is particularly evident when genomic sequencing studies yield ambiguous findings, such as variants of uncertain significance (VUSs) in syndromic genes.1 Moreover, characteristic phenotypic features, or “phenotypic handles,” often guide focused investigations. Rare phenotypically heterogeneous disorders present a considerable challenge in this respect due to prior ascertainment biases and limited cohort sizes.
Zhu-Tokita-Takenouchi-Kim (ZTTK) syndrome (OMIM #617140) is a rare phenotypically heterogeneous disorder characterized by developmental delay and a wide array of congenital multisystem anomalies. Haploinsufficiency of SON, mediated by either truncating variants or whole-gene deletions, has been identified as the underlying cause of ZTTK syndrome.2-5 SON encodes the RNA-binding protein SON and is constitutively expressed, though expression is enriched during early brain development.3 SON has been implicated in the splicing of pre-mRNA and has been shown to have a pivotal role in the regulation of pluripotency, cell-cycle progression, neurogenesis, and transcriptional regulation through its DNA-binding activity.6-10
The phenotypic spectrum of ZTTK syndrome has been highlighted in descriptions of >60 individuals.2-5,11-14 The core neurologic phenotypes identified include moderate-to-severe intellectual disability, hypotonia, and epilepsy. Brain MR imaging, available for 41 published patients, typically demonstrate a range of nonspecific structural abnormalities.12 Ventriculomegaly (24/41) is most frequently observed, with cortical dysplasia (7/41) and abnormalities of the corpus callosum (18/41), cerebellum (6/41), and cerebral WM (6/41) also being noted.12 Growth retardation and neonatal feeding difficulties are frequently reported. Dysmorphic facial features are observed in all cases; however, these features are variable, with no consistent pattern observed.12 Hearing and visual impairments as well as a range of cardiac, gastrointestinal, musculoskeletal, and renal abnormalities are common. The phenotypic features associated with ZTTK syndrome are both nonspecific and highly variable, presenting a diagnostic challenge.
Here we report 7 unrelated individuals with a confirmed molecular diagnosis of ZTTK syndrome. All individuals have periventricular nodular heterotopia (PVNH), a distinctive phenotypic feature on brain MR imaging, which we propose is an underappreciated element of ZTTK syndrome. PVNH is a neuronal migration disorder characterized by heterotopic GM nodules abutting the margins of the lateral ventricles. PVNH is an infrequent feature of many rare genetic disorders, with a small number manifesting it as a consistent association, as summarized by Vriend and Oegema.15 The major genetic locus for PVNH is FLNA, with pathogenic variants estimated to account for one-quarter of cases.16
In addition to PVNH, we observed a previously unrecognized characteristic pattern of additional structural brain anomalies in ZTTK syndrome. These include abnormalities of the pituitary gland, caudate nuclei, and hippocampi and cerebral sulcation and gyrification. Our characterization of brain imaging findings in ZTTK syndrome will improve diagnostic formulation, particularly in circumstances in which genetic sequencing studies are not possible or identify a VUS in SON.
MATERIALS AND METHODS
Cohort Recruitment and Ethics
A cohort of 7 unrelated individuals with pathogenic variants in SON was assembled by 2 approaches. Individuals 1–5 were ascertained through a physician-initiated referral into a high-throughput sequencing study of individuals with PVNH. This cohort was pre-emptively screened for pathogenic variants in FLNA. Individuals 6 and 7 were identified through diagnostic sequencing studies and were included after demonstration of a pathogenic SON variant. Individuals 1–3 were previously analyzed by Heinzen et al,17 however, the pathogenic SON variants described here failed validation but are now confirmed as true-positives. Individuals 3 and 4 were reported in brief by Dingemans et al.12 This study was approved by the Health and Disability Ethics Committee of New Zealand (13-STH/56).
Sequence Analysis
Genomic DNA from probands and parents was extracted from peripheral blood for either whole-exome sequencing (WES) or whole-genome sequencing (WGS), summarized in Table 1. WES libraries were constructed for individuals 1–3 by Otogenetics (Atlanta) and sequenced using a HiSeq 2500. WGS libraries were prepared for individuals 4 and 5 using a TruSeq DNA Nano v2.5 kit and sequenced using a HiSeq X Ten at the Kinghorn Center for Clinical Genomics (Sydney). Paired-end FASTQ reads were aligned to the human reference GRCh37 using BWA MEM (Version 0.7.17; https://github.com/bwa-mem2/bwa-mem2) and outputted in BAM format.
Molecular findings identified in a cohort of 7 unrelated individuals with ZTTK syndrome
Single sample variant calls were produced using GATK HaplotypeCaller (Version 3.8; https://github.com/broadinstitute/gatk), in genomic VCF format. Genomic VCFs were then jointly genotyped using GATK GenotypeGVCFs producing a multisample VCF. Annotation with gene context information was performed using SnpEff (Version 4.3S; https://github.com/pcingola/SnpEff), and additional information from the gnomAD project (Version 2.1.1; https://gnomad.broadinstitute.org/)18 was added using BCFtools annotate (Version 1.9; https://github.com/samtools/bcftools).
Variant Prioritization
Sequence data from individuals 1–5 were filtered under a monogenic disease model. Variants were required to have a read depth of >7 and an alternative allele percentage of >20%. Missense and predicted loss-of-function variants were selected. Common variants with a gnomAD allele frequency of >0.001 were removed.
For individuals 1–4, parental sequence data were available, permitting identification of de novo variants. For individual 5, parental data were unavailable, so a restrictive disease-gene filtering model was implemented. Variants in genes with a dominant disease phenotype were retained based on data from the Developmental Disorder Genotype-Phenotype database.19 Remaining variants for all 5 individuals were interpreted on the basis of the American College of Medical Genetics and Genomics guidelines.1
Neuroradiologic Evaluation
Brain MR images were systematically reviewed by a pediatric neuroradiologist (S.M.) with >20 years of experience in the imaging of brain dysmorphology. Normative data for the corpus callosum20 and cerebellum21 were used to assess abnormalities of these structures.
Data Availability
Individual genomic data sets are not freely available due to restrictions imposed by the consenting process and privacy concerns.
RESULTS
Molecular Findings
De novo loss-of-function SON variants were identified for individuals 1–4, consistent with pathogenic class 5 variants causative of ZTTK syndrome.3 For individual 5, a heterozygous SON frameshift variant (NM_138927.2:c.5753_5756del) of unknown inheritance was identified, which was classified as a class 5 pathogenic variant based on recurrence in other published cases of ZTTK syndrome.3,4 Identified SON variants for individuals 1–5 were validated using polymerase chain reaction and Sanger sequencing.
Class 5 pathogenic SON variants were identified for individuals 6 and 7 before recruitment into this study. A recurrent heterozygous frameshift variant (NM_138927.2:c.3852_3856del)3 of unknown inheritance was identified for individual 6 and a de novo heterozygous whole-gene deletion (NC_000021.8:g.(34849772_34911445)_(35109752_35171289del) was identified for individual 7. Sequencing results are summarized in Table 1.
Clinical Descriptions
Individual 1.
Individual 1 is a female who presented with hypotonia and mild global developmental delay at 10 months of age. Intrauterine growth restriction occurred during pregnancy. Delivery was by cesarean delivery, and her weight and height were both below the 3rd centile. Poor feeding and febrile seizures were noted, with hemiplegic migraine attacks occurring from 6 years of age. She was diagnosed with mild intellectual disability in childhood. She had a ridged, sagittal suture, broad nasal bridge, anteverted nares, a small chin, and cupped posteriorly rotated ears.
Recurrent respiratory and middle ear infections occurred throughout childhood. At 13 years of age, she developed end-stage polyuric renal disease, requiring renal transplantation. When last examined at 17 years of age, her weight and head circumference were in the 50th centile, with height below the 3rd centile. She had hearing loss, hypermetropia, and convergent strabismus. Echocardiogram findings were normal.
Individual 2.
Individual 2 is a female who presented in early childhood with global developmental delay with severe intellectual disability and hypotonia. She had an irregular whole-body tremor from birth. Poor feeding occurred from early infancy, requiring the insertion of a gastrostomy tube at 6 years of age. Myoclonic seizures with fever began at 6 years of age, developing into multifocal myoclonic and clonic seizures by 11 years of age. At 13 years of age, she had an episode of status epilepticus. Dysmorphic features included a flat nasal bridge, broad nose, and anteverted nares. Divergent strabismus and nonsensorineural deafness were also noted.
When last examined at 28 years of age, her height was below the 1st centile, her weight was below the 3rd centile, and her head circumference was between the 2nd and 50th centile. She could walk with assistance but was nonverbal and displayed self-harm behaviors. Partial second- and third-digit syndactyly was noted. She had bilateral hypoplastic kidneys and osteopenia. She developed heart failure after an episode of sepsis.
Individual 3.
Individual 3 is a male who presented with global developmental delay with moderate intellectual disability. He had asthma and recurrent respiratory infections throughout infancy. Through childhood, he developed selective mutism and disrupted sleep patterns, as well as epilepsy with mixed seizure types. Major feeding difficulties occurred throughout his life, and he is now predominantly fed by gastrostomy.
During childhood, his height tracked below the 2nd centile, and his weight, between the 9th and 25th centiles. His dysmorphic features included down-slanting palpebral fissures and epicanthal folds. Craniosynostosis of the left coronal suture was observed. Other congenital anomalies included astigmatism, joint hypermobility, and a unilateral multicystic kidney.
Individual 4.
Individual 4 is a female who presented with moderate developmental delay and hypotonia. When last examined at 4 years of age, her height and weight were below the 1st and 3rd centiles, respectively. Dysmorphic features included down-slanting palpebral fissures, epicanthal folds, and a flat philtrum. Feeding difficulties were pronounced during infancy. Additional anomalies included fourth and fifth digit camptodactyly, sensorineural hearing loss, hypermetropia and astigmatism, strabismus, and a horseshoe kidney. She developed coronal craniosynostosis, requiring surgical intervention. After the operation, she developed seizures, well-controlled with levetiracetam.
Individual 5.
Individual 5 is a female who presented with moderate developmental delay with intellectual disability and hypotonia in early childhood. Notable facial features included frontal bossing and epicanthal folds. Conductive hearing impairment and strabismus were noted. She required surgical intervention for right-hip dysplasia. When last examined at 5 years 8 months of age, her height was at the 25th centile, her weight, in the 3rd centile, and her head circumference, in the 90th centile.
Individual 6.
Individual 6 is a male who presented with suspected seizures after several episodes of confusion, later diagnosed as a concussion. He was delivered at term after an uneventful pregnancy. Early childhood developmental milestones were normal, but he was diagnosed with mild intellectual disability during his early school years. When examined at 14 years of age, his height and weight were between the 10th and 25th centiles. No dysmorphic facial features were noted, but he had fifth finger and toe clinodactyly as well as elbow extension limitation. A ventricular septal defect was noted on an echocardiogram.
Individual 7.
Individual 7 is a female who presented prenatally with intrauterine growth restriction, breech presentation, and oligohydramnios. She was delivered at 37 weeks by cesarean delivery, with an 11th centile birth weight. During infancy she had central hypotonia with a peripheral increase in tone. At 4 years of age, she had onset of seizures, with an episode of status epilepticus. Her development was globally delayed, with suspected moderate intellectual disability and autism spectrum disorder.
During early childhood, her height and weight were between the 10th and 25th centiles, with a head circumference at the 73rd centile. She had down-slanting and short palpebral fissures, epicanthal folds, smooth philtrum, small mouth with thin lips, and low-set ears with diagonally creased earlobes. Additional anomalies included developmental hip dysplasia and a ventricular septal defect.
Neuroradiologic Findings
A prior diagnosis of PVNH was available for individuals 1–5 before recruitment into this study. The initial goal of neuroradiologic re-evaluation was to categorize the PVNH of these individuals and assess whether it demonstrates characteristics distinctive to ZTTK syndrome. However, after re-evaluation of MR images, PVNH was also identified for individuals 6 and 7, who were recruited solely on the basis of their molecular diagnosis of ZTTK syndrome.
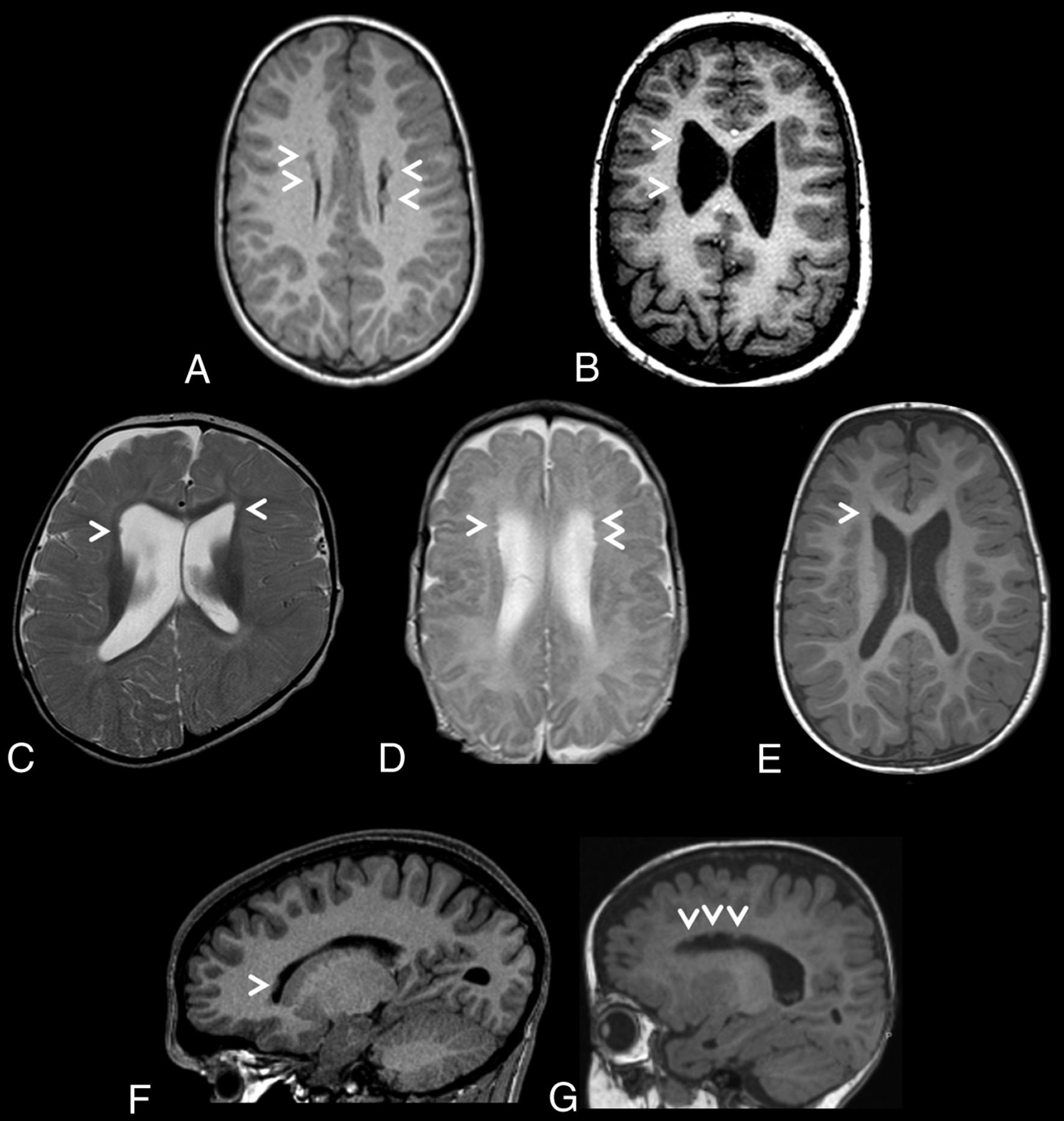
Heterotopic nodules were sparse, small, and predominantly frontal in all 7 individuals. They were bilateral in 5 of 7 cases. Similarly, sparse and small nodules were identified in the posterior infrasylvian regions in 3 of 7 cases. MR images depicting the PVNH distribution for each individual are presented in Fig 1. No nodules were observed in the occipital horns.

Brain MR images depicting periventricular nodular heterotopia distributions in a cohort of 7 unrelated individuals with ZTTK syndrome. A, Axial T1-weighted MRI of the brain of individual 1 at 7 years 7 months of age revealing sparse bilateral frontal heterotopia (white arrowheads). B, Axial T1-weighted MRI of the brain of individual 2 at 11 years and 6 months of age revealing sparse right unilateral frontal and midbody heterotopia (white arrowheads). C, Axial T2-weighted MRI of the brain of individual 3 at 1 year of age revealing sparse bilateral frontal horn heterotopia (white arrowheads). D, Axial T2-weighted MRI of the brain of individual 4 at 1 year 4 months of age revealing sparse bilateral frontal horn heterotopia (white arrowheads). E, Axial T1-weighted MRI of the brain of individual 5 at 1 year 8 months of age revealing a heterotopion of the right frontal horn (white arrowhead). F, Right parasagittal T1-weighted MRI of the brain of individual 6 at 15 years 2 months of age revealing a heterotopion of the right frontal horn (white arrowhead). G, Left parasagittal T1-weighted MRI of the brain of individual 7 at 1 year 2 months of age revealing sparse left frontal horn heterotopia (white arrowheads).
In addition to PVNH, consistent involvement of abnormalities in specific structures were identified in all 7 individuals, summarized in Table 2. These included malformations of the corpus callosum, cerebellum, pituitary stalk, caudate nuclei, hippocampi, and cortex.
Neuroradiologic findings identified in a cohort of 7 unrelated individuals with ZTTK syndrome
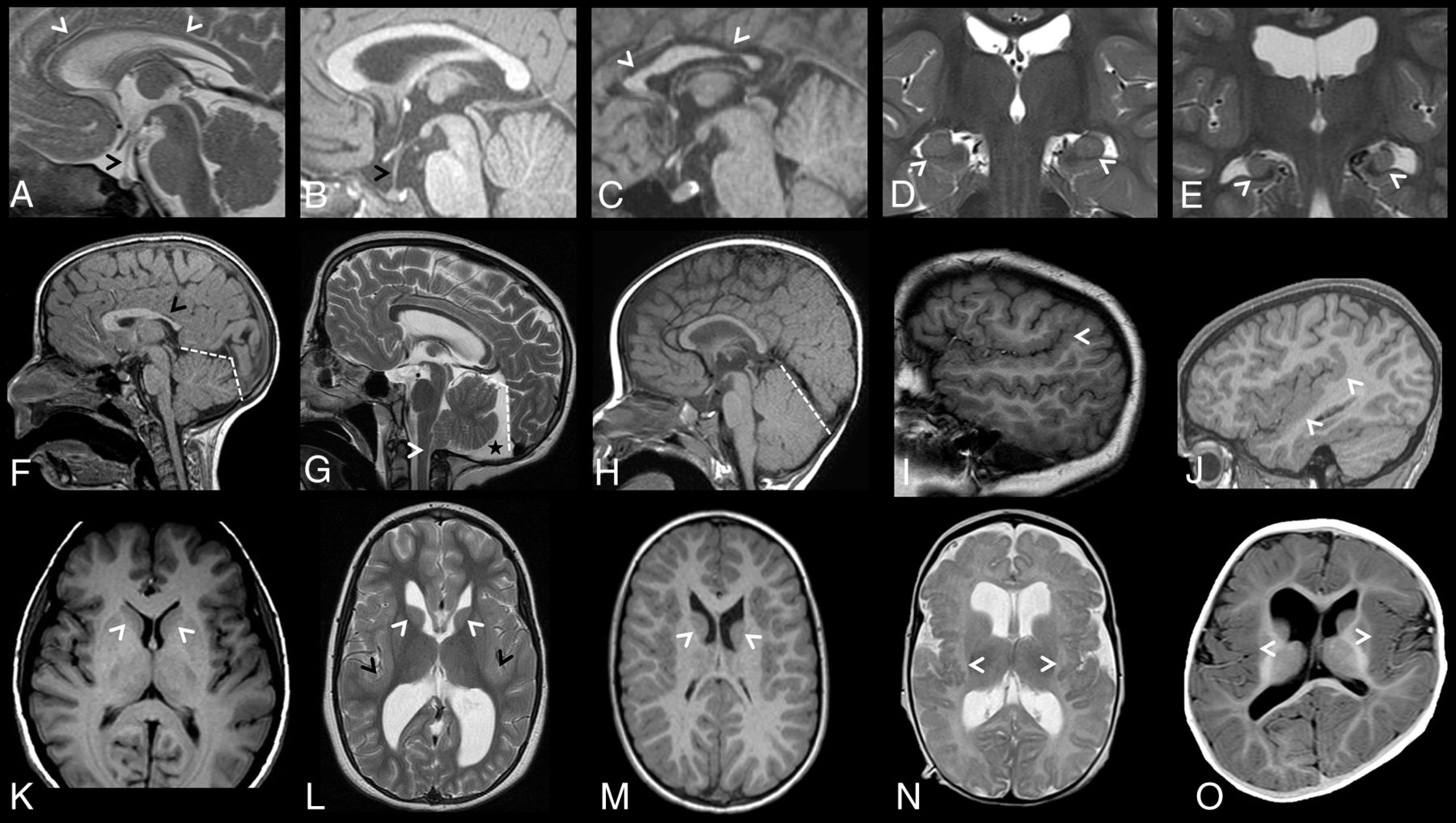
Corpus callosum abnormalities were frequently observed and included short anterior-posterior diameter (<3 centiles, 3 of 7) and thinning of the corpus callosum, particularly at the genu (<3 centiles, 5 of 7).20 Dysmorphic corpora callosa were observed in 2 individuals, depicted in Fig 2C, -F. Abnormalities of the pituitary gland and stalk were also seen, with glands appearing small (5 of 7), with a markedly elongated pituitary stalk (5 of 7). The pituitary stalk elongation observed in individuals may relate to platybasia or a similar anomaly at the base of the skull; however, this was not assessed in the current study. Examples of identified corpus callosum and pituitary abnormalities are depicted in Fig 2A–C.

Brain MR images depicting additional brain anomalies in a cohort of 7 unrelated individuals with ZTTK syndrome. A, Sagittal midline T2-weighted MRI of the brain of individual 4 at 1 year 4 months of age showing a thin corpus callosum (white arrowheads), an elongated pituitary stalk (black arrowhead), and small pituitary gland. B, Sagittal midline T1-weighted MRI of the brain of individual 5 at 1 year 8 months of age showing an elongated pituitary stalk (black arrowhead) and a small pituitary gland. C, Sagittal midline T1-weighted MRI of the brain of individual 6 at 15 years 2 months of age showing a shortened corpus callosum with a hypoplastic anterior and posterior body (white arrowheads), a normal-sized pituitary stalk, and a normal-sized pituitary gland. D, Coronal T2-weighted MRI of the brain of individual 5 at 1 year 8 months of age revealing normal-sized hippocampi with a lack of internal architecture (white arrowheads). E, Coronal T2-weighted MRI of the brain of individual 2 at 11 years 6 months of age revealing small malrotated hippocampi (white arrowheads). F, Sagittal midline T1-weighted MRI of the brain of individual 1 at 7 years 7 months of age showing a box-shaped posterior fossa (white dashed lines) and a shortened corpus callosum with a hypoplastic posterior body, isthmus, and splenium (black arrow). G, Sagittal midline T2-weighted MRI of the brain of individual 2 at 11 years 6 months of age showing a box-shaped posterior fossa (white dashed lines) with a posterior fossa cyst (black star) and dysmorphic cerebellar tonsils (white arrowhead). H, Sagittal midline T1-weighted MRI of the brain of individual 3 at 1 year of age shows a large cerebellum. I, Right parasagittal T1-weighted MRI of the brain of individual 2 at 11 years 6 months of age revealing perisylvian polymicrogyria with upswept Sylvian fissures (white arrowhead). J, Right parasagittal T1-weighted MRI of the brain of individual 6 at 15 years 2 months of age revealing a thick perisylvian cortex and dysgyria (white arrowheads). K, Axial T1-weighted MRI of the brain of a control individual depicting caudate nuclei with caudate heads (white arrowheads) that are less rounded than in L and M. L, Axial T2-weighted MRI of the brain of individual 2 at 11 years 6 months of age revealing a dysmorphic caudate nucleus with rounded and malrotated caudate heads (white arrowheads), large dysmorphic frontal and occipital horns, and a bilaterally thickened perisylvian cortex (black arrowheads). M, Axial T1-weighted MRI of the brain of individual 1 at 7 years 7 months of age revealing dysmorphic caudate nuclei with rounded and malrotated caudate heads (white arrowheads) and large dysmorphic frontal horns. N, Axial T2-weighted MRI of the brain of individual 4 at 1 year 4 months of age revealing bilateral posterior perisylvian polymicrogyria (white arrowheads). O, Axial T1-weighted MRI of the brain of individual 3 at 1 year of age revealing a thick perisylvian cortex and dysgyria (white arrowheads).
The cerebellum was large in all individuals, with 4 individuals above the 97th centile for craniocaudal vermis height and 6 individuals above the 97th centile for anterior-posterior vermis diameter.21 The cerebellar tonsils were herniated through the foramen magnum in 2 of 7 individuals (>3 mm below the level of the foramen magnum). For individual 4, MR images were available at 2 time points, revealing a normal-sized cerebellum at 16 days of age. However, by 16 months of age, the cerebellum was overgrown with tonsillar herniation. Appearances most consistent with posterior fossa cysts were observed in 3 of 7 individuals. The posterior fossa was “box-shaped” in 5 of 7 individuals due to a steepened tentorial angle and may be related to an apparent increase in cerebellar size in these individuals. Representative MR imaging slices showing posterior fossa abnormalities are presented in Fig 2F–H.
The caudate nuclei were dysmorphic in all individuals, with caudate heads appearing malrotated and globular in 5 individuals, depicted in Fig 2L, -M. Abnormalities in the lentiform nucleus were frequently observed, restricted to the globus pallidus in 3 individuals and generalized in an additional case. Abnormal hippocampi were seen in all individuals, appearing small and malrotated in 4 individuals (Fig 2E). The hippocampi of the remaining 3 individuals showed a lack of internal architecture (Fig 2D). The thalami were normal in all individuals.
The lateral ventricles were dysmorphic and dilated in all individuals, affecting both the frontal (6 of 7) and occipital (3 of 7) horns. Further abnormalities of the ventricular system were also observed, with a small fourth ventricle in all individuals, likely secondary to enlargement of the cerebellum. A generalized decrease in WM volume was seen in 4 individuals, with delays in age-appropriate myelination in 3 individuals.
Malformations in cerebral cortical gyrification and sulcation were seen in all individuals. Examples are depicted in Fig 2I, -J, -L, -N, -O. These cortical malformations included bilateral perisylvian polymicrogyria (3 of 7), bilateral perisylvian dysgyria or polymicrogyria-like cortical dysplasia (2 of 7), and perisylvian cortex thickening (3 of 7). Disorganized sulcation was also observed in 2 individuals, appearing to be especially localized to the temporal lobe in individual 1.
DISCUSSION
ZTTK syndrome is a rare disorder with a wide phenotypic spectrum, posing diagnostic challenges in the absence of genomic testing. Instead, high-throughput sequence screening of SON has been suggested as the only practical way of diagnosing ZTTK syndrome.12-14 The cohort described here manifests the core phenotypic features of ZTTK syndrome, including developmental delay, seizures, and dysmorphic features. However, this core presentation remains both nonspecific and highly variable, offering limited utility for recognition in a clinical setting.
We identified PVNH as an imaging feature that may aid clinical recognition of ZTTK syndrome and analyzed a cohort of 7 cases with this phenotypic feature. The distribution of FLNA-associated PVNH overlaps that seen with the frontal predominant distribution of ZTTK syndrome. However, FLNA-associated nodules are typically larger and more contiguous and do not frequently extend to the temporal horns.16 In addition, the co-occurrence of polymicrogyria and other cortical malformations distinguishes the pattern observed in ZTTK syndrome from FLNA-associated PVNH. However, these features alone are not pathognomonic of this condition because similar combinations of features have been previously observed, such as in individuals with MAP1B- and NEDD4L-associated cortical malformations.17,22,23
Three additional instances of PVNH in ZTTK syndrome have been described previously, of a total of >41 individuals with MR imaging screening.3,11,12,24 This observation likely represents an underestimate of the true prevalence of PVNH in ZTTK syndrome because previous studies did not report PVNH as a discrete neuroradiologic entity. Additionally, the subtle nature of the sparse and isolated heterotopia identified here could lead to underreporting, as occurred for individuals 6 and 7.25,26
Neuroradiologic assessment of MR imaging data led to the identification of a set of brain structures with recurrent abnormalities. These features may suggest the diagnosis of ZTTK syndrome in situations in which hypothesis-free high-throughput sequencing is not available. Additionally, these features may support the pathogenic nature of VUS identified in SON, such as the multiple missense variants that have now been reported in conjunction with ZTTK syndrome-like features.4,11,12 Interestingly, craniosynostosis was also noted in 2 patients, both involving the coronal sutures. Craniosynostosis has been previously reported in individuals with ZTTK syndrome and could represent another phenotypic feature overrepresented in this disorder.3,11
Most individuals reported in this study were recruited based on the presence of PVNH with a subsequent molecular diagnosis, representing a potential recruitment bias. As a consequence, the neuroradiologic signature identified may be associated with PVNH, with no direct association with ZTTK syndrome. The frequency of these newly identified neuroradiologic features in ZTTK syndrome will be best assessed through studying individuals recruited purely on the basis of a molecular diagnosis. Re-assessing published ZTTK cohorts for these imaging features may validate the clinical utility of the observations made in this cohort.3,4,11,12
CONCLUSIONS
We identify a recurrent set of cortical malformations in a cohort of individuals with ZTTK syndrome. These imaging features have the potential to aid in the diagnosis of ZTTK syndrome in the absence of high-throughput sequencing.
Acknowledgments
Patients and their families are thanked for their willing participation. The authors wish to acknowledge access to New Zealand eScience Infrastructure high-performance computing facilities for the processing of whole-exome and genome sequence data in this research.
Footnotes
S.P. Robertson is supported by the Health Research Council and Cure Kids. B.J. Halliday is supported by a University of Otago Doctoral Scholarship. R.J. Leventer is supported by a Melbourne Children’s Clinician Scientist Fellowship. I.E. Scheffer is supported by the Australian National Health and Medical Research Council, the Australian Medical Research Future Fund, and the Australian Epilepsy Research Fund.
Disclosure forms provided by the authors are available with the full text and PDF of this article at www.ajnr.org.
References
- Received February 24, 2022.
- Accepted after revision August 8, 2022.
- © 2022 by American Journal of Neuroradiology





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.