
Abstract
Summary: Schwartz-Jampel syndrome is a rare, inherited disorder characterized by myotonia, skeletal deformities, facial dysmorphism, and growth retardation. In this report of an adolescent male patient with Schwartz-Jampel syndrome, CT and MR imaging revealed basilar invagination, platybasia, Chiari I malformation, hyperpneumatized mastoids with intramastoid dural sinuses, platyspondyly, bulbous zygoma, and blunted pterygoid processes.
Osteochondromuscular dystrophy or Schwartz-Jampel syndrome is an autosomal recessive disease characterized by muscle stiffness, mild muscle weakness, and various skeletal deformities including platyspondyly and kyphoscoliosis (1). The case reported herein illustrates clinically significant CT and MR imaging findings of the face and craniocervical junction in a patient with Schwartz-Jampel syndrome.
Case Report
We present the case report of a 15-year-old white male patient with type IB Schwartz-Jampel syndrome. Shortly after birth, the patient began experiencing severe muscle stiffness, joint deformities at the ankles and knees, and bowing of the legs. After perplexing his physicians for approximately 1 year, genetic testing and review of clinical history confirmed the diagnosis. Physical examination was notable for dysmorphic facies, including blepharophimosis with stiff facial musculature, kyphoscoliosis, and various extremity deformities, but showed intact head and truncal motor control. Because of progressive deformity of the right lower extremity, which prevented ambulation, the patient had undergone several corrective osteotomies of the proximal femur and proximal tibia. He also required excision of the uvula and tonsillectomy for obstructive sleep apnea and was expected to undergo spinal fusion for kyphoscoliosis. With the exception of genetic liability, he had no history of constitutional illness, such as immunocompromise. Showing no evidence of mental retardation, he exhibited normal affect and uplifting optimism despite his disability.
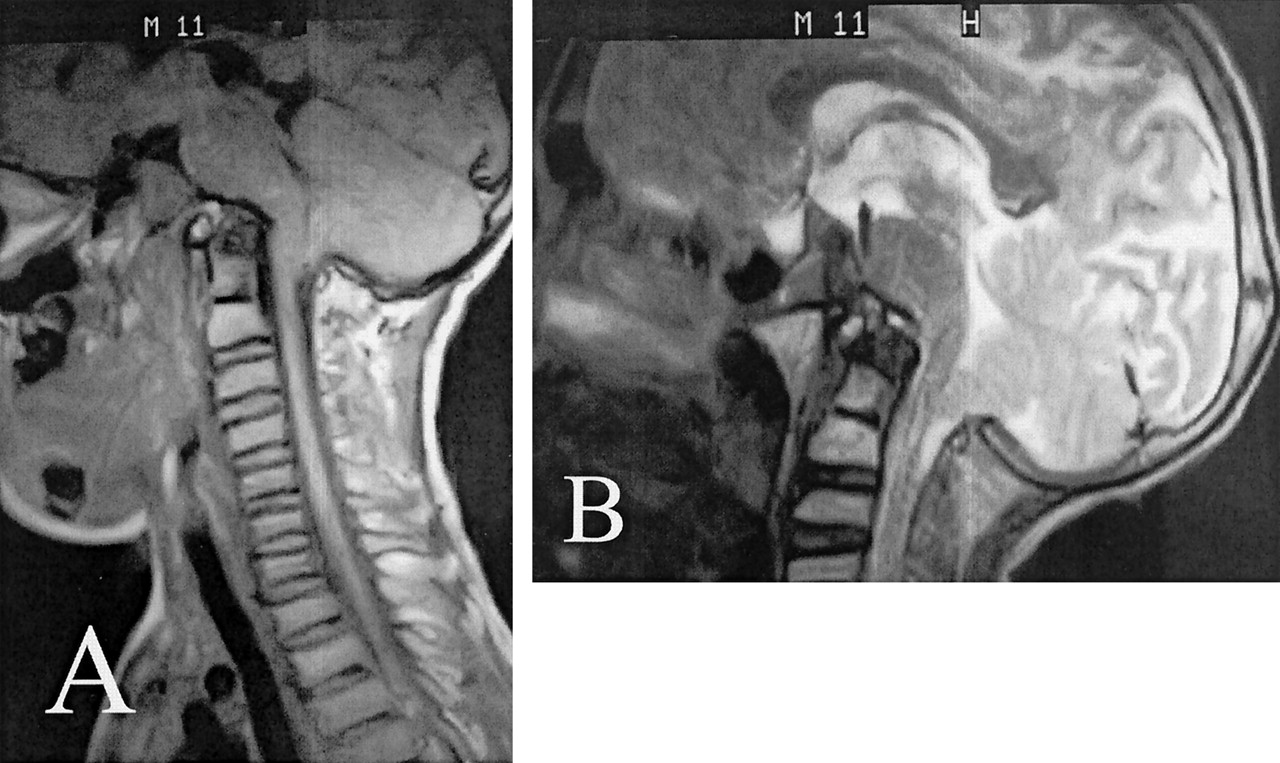
Unenhanced sagittal view MR imaging of the head and cervical spine (Fig 1A) revealed pronounced basilar invagination and narrowing of the sagittal diameter of the upper cervical spine and foramen magnum. In addition, the anterior ring of C1 was rostrally located posterior to the upper clivus on the CT scan (Fig 2A). The nasopharyngeal soft tissues were elevated superiorly (Fig 2B). Brain development and myelination appeared appropriate without cortical atrophy, callosal dysgenesis, or migrational anomaly. However, in association with the basilar invagination and platybasia, a hyperacute angle of the pontomedullary junction and a Chiari I malformation were noted on MR images (Fig 1B). Bulbous zygomatic processes of the maxillae and blunted pterygoid process plates were observed on CT (Fig 3).

Unenhanced sagittal view MR images obtained when the patient was 11 years old.
A, T1-weighted MR image shows platybasia and basilar invagination in conjunction with an associated hyperacute angle of the pontomedullary junction. A Chiari I malformation is seen, with tonsillar ectopia 7 mm below the opisthion-basion line. Multilevel, cervicothoracic platyspondyly is readily apparent.
B, T2-weighted MR image.

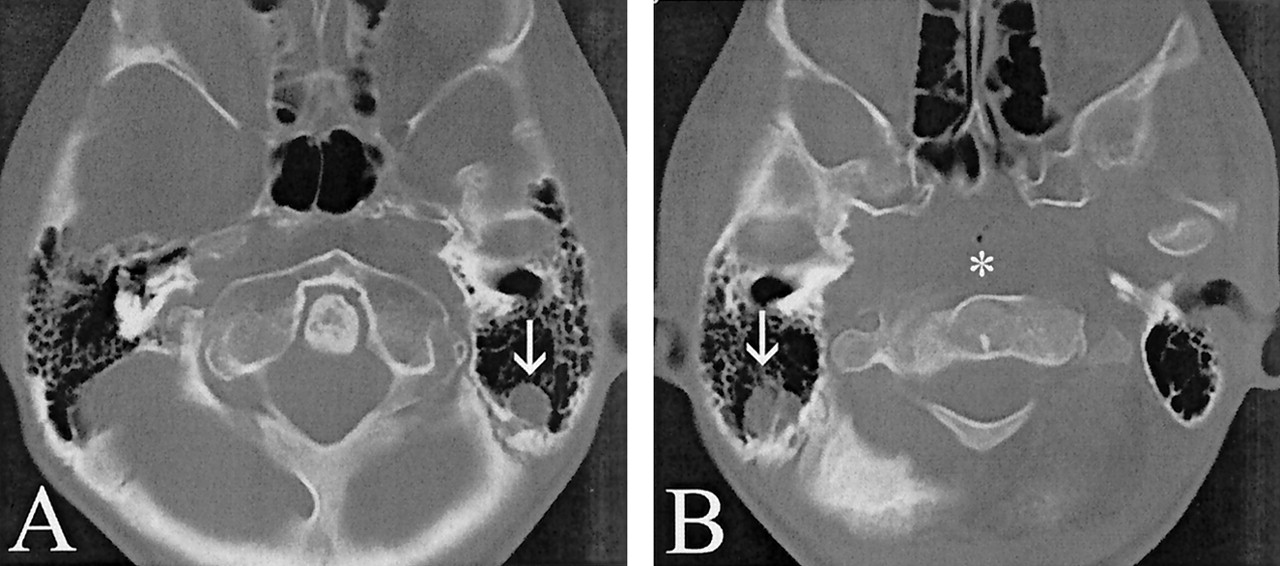
Axial view unenhanced CT scans obtained when the patient was 15 years old.
A, Basilar invagination is evidenced by the rostral location of the C1 ring posterior to the upper clivus. Hyperpneumatized mastoids are seen surrounding portions of the left sigmoid-transverse sinus junction (arrow).
B, Hyperpneumatized mastoids are seen surrounding portions of the right sigmoid-transverse sinus junction (arrow). Note the rostral location of the nasopharyngeal soft tissues (asterisk) relative to the orbits, mimicking a destructive process of the skull base.

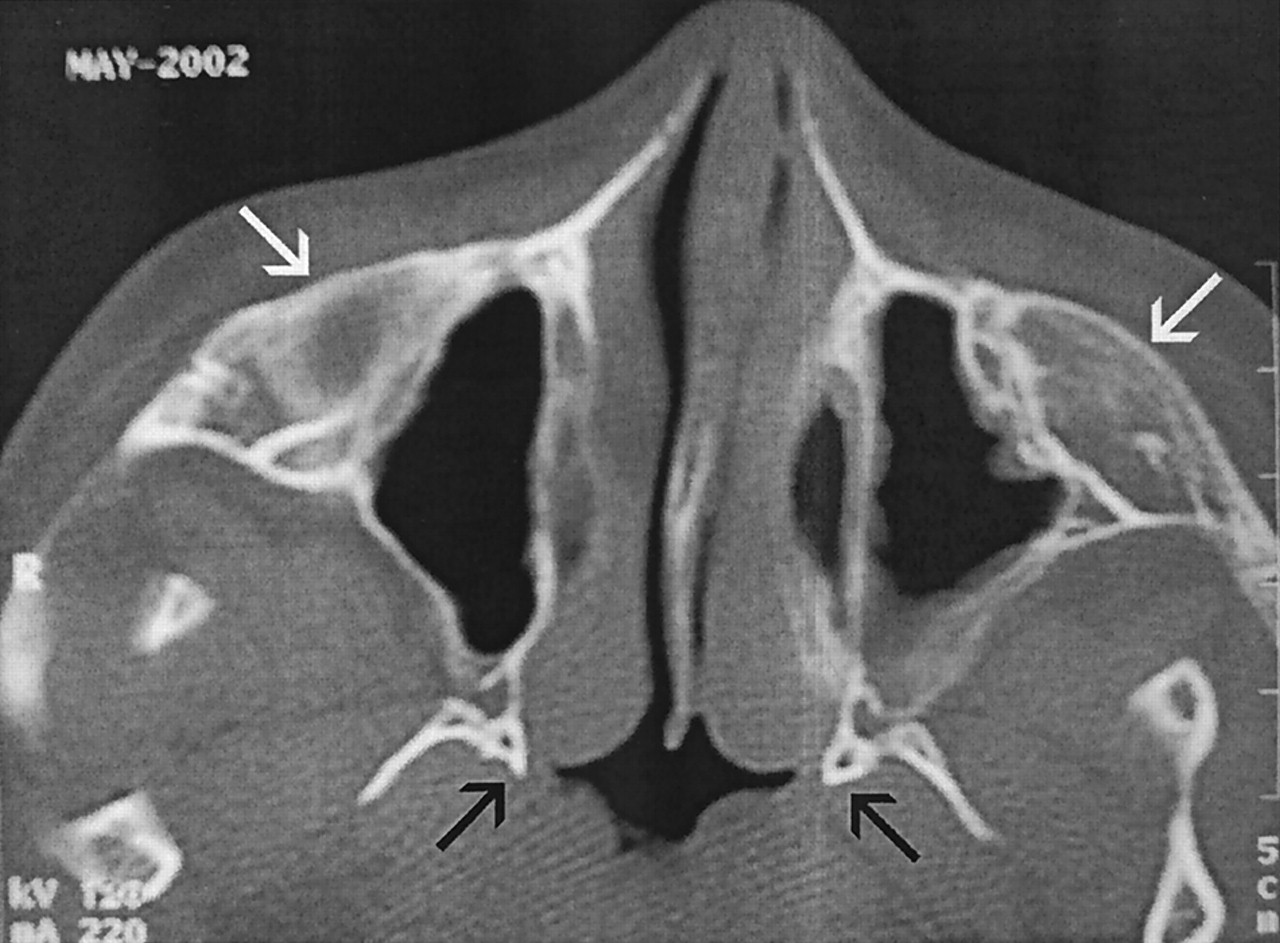
Axial view unenhanced CT scan obtained when the patient was 15 years old. Bulbous zygomatic processes (white arrows) of the bilateral maxillae and blunted pterygoid process medial plates (black arrows) are noted. Prominent nasopharyngeal soft tissues nearly obliterate the upper airway.
Discussion
First described in 1962 by Oscar Schwartz and Robert Jampel (1), Schwartz-Jampel syndrome can be categorized into types 1A, 1B, and 2. Type 1A presents later in childhood and is less severe than type 1B, which usually is apparent at birth and frequently has more prominent bone dysplasia (2). In types 1A and 1B, there does not seem to be a shortened lifespan, but there is significant morbidity associated with constant muscle stiffness and bone abnormalities (3, 4). Type 2 is likely Stuve-Wiedemann syndrome, typified by myotonia, contractures, severe long bone bowing, anesthesia-induced hyperthermia, increased infantile mortality with respiratory distress, and sporadic reports of cortical atrophy revealed by MR imaging and absent corneal reflexes (2, 5–8). Bone and joint deformities contribute to abnormal motor development, which can usually be seen during the first year of life (2). Bone abnormalities include kyphoscoliosis, platyspondyly with resulting dwarfism or diminished stature and a short neck, coxa valga, irregularities of the capital femoral epiphyses, bowing of the long bones, and pectus carinatum (2). In addition, patients exhibit a puckered facial appearance, narrow palpebral fissures (blepharophimosis), micrognathia, and hypertrichosis of the eyelids (2). The incidence of mental retardation in patients with Schwartz-Jampel syndrome has been estimated to be 25% (9). This autosomal recessive disorder is distinguished from other myotonic disorders by its prominent facial features, skeletal deformities, and associated dwarfism (2).
The pathophysiology of the disease is not clearly understood, but electromyography shows nonvariable, continuous high frequency electrical activity (10). Studies have speculated that a mutation in the gene responsible for perlecan, a large heparan sulfate proteoglycan, may be responsible (11). In 1995, the Schwartz-Jampel syndrome gene was mapped to chromosome 1p (locus 1p34-p36.1) in several inbred families of different ethnicity (12). Other genetic or in utero factors may be involved; phenotypical variability has been noted among affected family members.
Approximately 85 cases of Schwartz-Jampel syndrome have been reported with the overwhelming majority of publications addressing the genetic considerations and the imaging abnormalities of the appendicular skeleton and spine. The literature regarding conventional radiology of Schwartz-Jampel syndrome describes the many osseous abnormalities that range from pelvic deformity and irregularities in the long bones to scoliosis, platyspondyly, and deficient ossification centers or coronal clefts in anterior vertebral bodies (13). CT has documented abnormally high attenuation in the sternocleidomastoid muscles and low attenuation in the paraspinal, quadriceps, sartorius, soleus, and gastrocnemius muscles (14). Cranial sonography found corpus callosal agenesis, a prominent third ventricle, and a large cyst of the septum pellucidum in one patient who displayed features of both Schwartz-Jampel syndrome and Stuve-Wiedemann syndrome (15). In a separate case of Stuve-Wiedemann syndrome, cortical brain atrophy was noted on MR images (7). Of note, the results of cross-sectional imaging (CT of the head, two patients; transcranial sonography, one patient) of three patients with type 2 Schwartz-Jampel syndrome (ie, Stuve-Wiedemann syndrome) were reported to be normal according to a study by Al-Gazali et al (16). In a separate publication illustrating a 16-year-old female patient with Schwartz-Jampel syndrome and compressive myelopathy, platybasia and basilar impression of the skull were shown on plain roentgenograms whereas myelography showed narrowing of the cervical sagittal diameter from the third to sixth cervical “roots” and dorsal displacement of the cord (17). Overall, no clear pattern has emerged in classifying the neuroradiologic findings of patients with Schwartz-Jampel syndrome or Stuve-Wiedemann syndrome.
Conclusion
Schwartz-Jampel syndrome is a rare disease with much variability and degree of severity. The craniocervical MR imaging and CT findings presented might represent an unrecognized subtype or variant of Schwartz-Jampel syndrome. Further reports of Schwartz-Jampel syndrome and the associated cross-sectional imaging findings should assist in furthering the understanding of, and subclassification within, this inherited disease.
References
- Received December 31, 2002.
- Accepted after revision March 25, 2003.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.