
Abstract
SUMMARY: Histologically identified intracellular and extracellular inclusions and structures often provide a tissue diagnosis of a specific disease process. Moreover, these deposits may provide clues about the pathogenesis of the disease in which they are found. Two distinctive structures seen within the brains of patients clinically diagnosed with dementia of the Alzheimer type are extracellular plaques and intracellular neurofibrillary tangles. The purpose of this report is to review the significance of plaques and neurofibrillary tangles in the context of Alzheimer disease.
Histologically identified cellular deposits often provide a tissue diagnosis as well as clues about the pathogenesis of the disease that they represent. Two distinctive structures seen within brains of patients with dementia of the Alzheimer type (DAT) are plaques and tangles. Appreciation of these tissue inclusions enhances the interactions of the neuroradiologist with the neuropathologist, neurologist, geriatric psychiatrist, and geriatrician and deepens the understanding of Alzheimer disease (AD), the all-too-common neurodegenerative disorder. Moreover, neuroradiologists are likely to play important roles in the future regarding the detection and differential diagnosis of AD, given the increasing prevalence of DAT in the aging population and promising research aided by such tools as positron-emission tomography (PET), quantitative structural imaging, molecular imaging, diffusion tensor imaging, and functional MR imaging. The purpose of this report is to review the significance of plaques and tangles in AD.
Background
In November 1901, a 51-year-old woman living in Frankfort, Germany, experienced increasing confusion, paranoia, and delusions and was admitted to the local asylum. There she was examined by the young psychiatrist Alois Alzheimer (1864–1915).1–3 Auguste D., as she was named in various reports, was fascinating in that her obvious dementia and complicating psychosis occurred at a much earlier age than the often-observed dementia of the elderly and that her deterioration progressed relatively rapidly.4 Following her hospitalization, Alzheimer left Frankfurt to join the famed pioneer of psychiatric taxonomy, Emil Wilhelm Magnus George Kraepelin (1856–1926), at the Psychiatry Department of Heidelberg University. Both physicians subsequently moved to Munich, where Kraepelin assumed the chair of Psychiatry at the Ludwig-Maximilians-Universität and built an internationally recognized program noted for the groundbreaking concept of biologic causation and neuropathologic correlation of disease presenting as psychiatric illness.5,6 When Auguste D. died in 1906, her brain was sent to Alzheimer in Munich for dissection at his request. He would present his findings at the 37th annual meeting of Southwest German psychiatrists in Tübingen, Germany, in November of that year, and would publish the account in 1907.7 His terse but extraordinarily prescient observations would launch a century of investigation into the causes of this early-onset dementia.
In that report, Alzheimer described miliary foci of extracellular structures, which would later be known as “plaques” as well as previously unrecognized intracellular flame-shaped fiberlike bundles that would be named “tangles.”7,8 Alzheimer was a fastidious observer and excellent microscopist, and his descriptions are still regarded as highly accurate. Alzheimer's student and colleague, Gaetano Perusini (1879–1915), would later compile 4 cases, including Alzheimer's original patient, and would more fully outline the clinical and pathologic findings.9,10 By 1910, Alzheimer had extensively studied the 3-year downhill course of a second patient, Johann F., a 56-year-old day laborer. Notably, this patient did not demonstrate tangles.1,11,12 Convinced that the early onset distinguished these patients, Kraepelin honored his protégé by naming the presenile dementing disorder “Alzheimer disease” in the eighth edition of his then-standard psychiatry textbook.5,13–16 Alzheimer's contribution to knowledge about presenile dementia resides in his identification of the clinical and pathologic features of the disorder, which, in turn, contributed to the recognition that dementia was an illness rather than an expected phenomenon of aging. Refinement of pathologic techniques some decades later would rekindle interest in this disorder.
What Are Plaques?
Alzheimer's first foundational observation was the presence of small spheric structures within the cerebral cortex, measuring 10–160 μm in diameter, which he termed “drusen” or “plaques.”17,18 Alzheimer was not the first investigator to describe cerebral plaques, though he was the first individual to record their presence in the presenile form of dementia. Several earlier researchers had noted similar inclusions in the cortex of autopsied elderly patients with dementia.17,19–22 Even the acclaimed Czech neurologist and psychiatrist, Arnold Pick (1851–1924), Kraepelin's archrival and chief psychiatrist at the University of Prague, who is now known for the frontotemporal subtype of dementia that bears his name, had recognized focal abnormalities in the cortex of an elderly patient.3
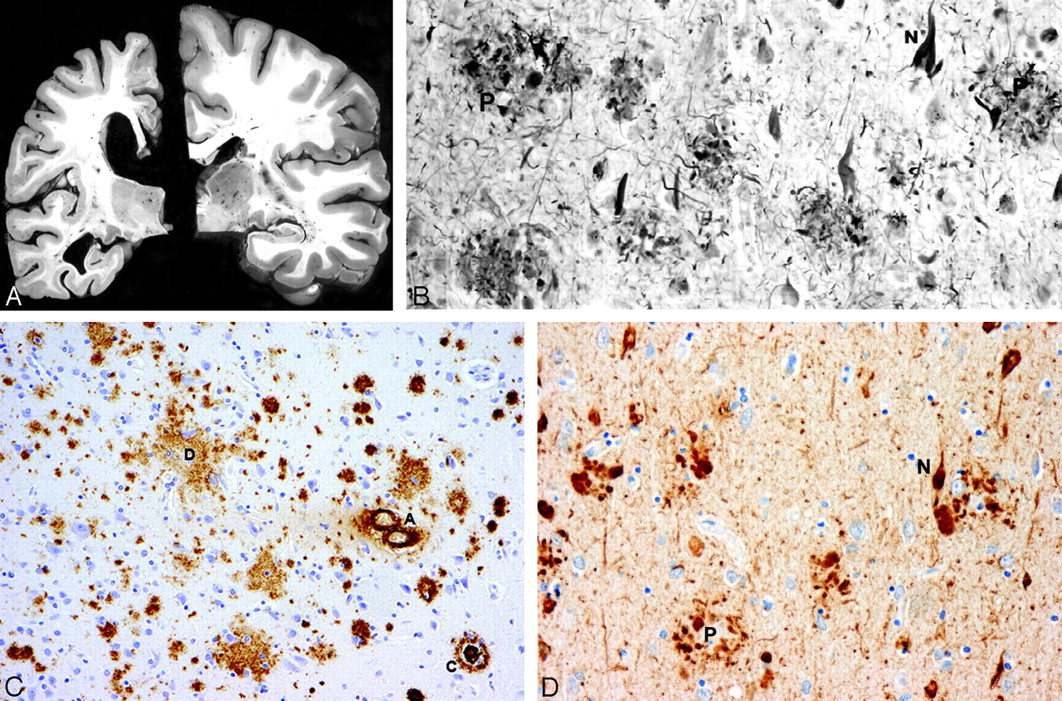
Plaques are proteinaceous extracellular deposits consisting primarily of amyloid-β (Aβ) peptide fragments. The most abundant species of Aβ found in plaques are either 40 or 42 amino acid residues in length (Fig 1).18,21,23,24 The β designation is derived from the secondary structure of the constituent protein in which parallel strands of amino acids are laterally associated with each other due to hydrogen bondings to form a sheetlike configuration of protein known as a β-pleated sheet. Curiously, the name “amyloid” (derived from Latin “amylum” or starch), as coined by the famous German pathologist and statesman, Rudolph Ludwig Karl Virchow (1821–1902), is a misnomer, because he thought the protein was actually a starchlike compound, due to its behavior with periodic-acid-Schiff stain.25 In addition to the Aβ peptide, plaques also contain a variety of other components such as apolipoprotein E, α-1-antichymotrypsin, and proteoglycans.18,24

A, Atrophy of the brain. On the left, a section of the hemibrain of a 70-year-old patient with AD and, on the right, a healthy aged control brain. The AD brain shows marked atrophy, dilation of the lateral ventricle, and a small hippocampus. B, Neurofibrillary tangles (N) and neuritic plaques (P) in the hippocampus. Modified Bielschowsky silver impregnation. C, β-amyloidosis in the frontal lobe: a diffuse plaque (D), a cored plaque (C), and cerebral amyloid angiopathy (A). β-amyloid (10D5) immunohistochemistry. D, Neurofibrillary tangles (N) and neuritic plaques (P) in the frontal lobe. Phosphorylated ô immunohistochemistry.
Associated with many of the plaques is evidence of inflammation and abnormal neuronal processes called dystrophic neurites.24 Fibrous astrocytes demonstrating increased amounts of glial fibrillary acidic protein (GFAP) mark this inflammatory response.18,26 GFAP is a cytoskeletal intermediate protein filament involved in glial cell adhesion, motility, and shape and tends to be upregulated in stress and injury.27–30 Fibrous astrocytes also appear in regions of neurofibrillary tangles (NFTs) and neuronal loss.18 Additionally, activated microglia surround plaques. They likely participate in pathogenesis, but their exact role remains unclear.18
The dystrophic neurites, consisting of swollen axons and dendrites containing lysosome-related attenuated bodies, immunologically stain for chromogranin A and ubiquitin. Chromogranin A is a secretory protein found in neurons and endocrine cells, and ubiquitin is a protein that regulates nonlysosomal degradation of other proteins.27,31–33 Some of these neurites may also contain paired helical filaments, which are strands of an aggregated protein known as τ, wound in a leftward configuration, repeating every 80 nm.24,34,35
Several types of plaques have been described. Diffuse plaques are focal poorly marginated collections of aggregated Aβ peptide that are not fibrillar and that lack dystrophic neurites, glial reaction, or any organized internal architecture.18,36 Neuritic plaques contain an attenuated central core of fibrillar Aβ peptide and have neighboring dystrophic neurites.18,24,37 They are associated with the degeneration observed at the synaptic junction and are surrounded by reactive astrocytes and activated microglial cells.24,37 Neuritic plaques also frequently are associated complement factors and immunoglobulins and a surrounding halo that includes fiberlike structures.37,38 Finally, so-called burnt-out plaques represent an end stage process and feature a condensed amyloid core without associated neurites.24 Some investigators contend that the stages of plaque development and resultant neuronal damage begin with the formation of the diffuse plaque, which then advances to the neuritic variety and eventually concludes with the burnt-out type.24,39 There is, however, no direct evidence that an evolution of plaque formation occurs.
Plaques occur abundantly in regions of the association areas of the neocortex, posterior cingulate, and limbic cortex.18 With the progression of the disease, plaque attenuation increases, with only late involvement of the primary sensory and motor areas.40
What Are Tangles?
Although plaques had been previously recognized, Alzheimer's novel observation was the presence of intraneuronal NFTs (“Fibrillenbundel”).7,17 These tangles resemble condensed serpentine fibrils and are primarily composed of τ protein arranged in hyperphosphorylated paired helical strands (Fig 1).23,34,41 Normal τ protein promotes the assembly of microtubules, which, in turn, help organize the 3D architecture of the neuron and assist in the transportation of protein and enzyme-containing vesicles essential in cell maintenance and function.42 For unclear reasons, τ protein aggregates inside neurons and their processes, and microtubules tend to depolymerize. This process is associated with hyperphosphorylation of τ protein and likely disrupts normal microtubule function. In addition, aggregated τ protein itself may lead to neuronal dysfunction.23,43 The cause for the abnormal phosphorylation is unclear, but altered function of several protein kinases or phosphatases or a maladaptation due to cellular or microenvironmental stress may be implicated.44,45
Initially, the τ protein is dispersed within the neurons. As it aggregates into defined filaments, silver staining reveals the classic NFTs within the neuronal cell bodies and dendrites. With the death of the neurons, the NFTs remain as extracellular “ghost tangles” or “tombstones.”24 The NFTs demonstrate a consistent pattern of involvement that may be useful for staging the disease in autopsied brain specimens.18,40,46,47 Patients with preclinical DAT have significant entorhinal cortex involvement with progression to the limbic cortex and eventually the isocortex as the disease clinically advances.40,46,47 However, τ pathology is not restricted to DAT. NFTs have also been identified in neurons, astrocytes, and oligodendrocytes of progressive supranuclear palsy, corticobasal degeneration, Pick disease, Parkinson dementia complex of Guam, and certain forms of familial frontotemporal dementia. Apparently, different patterns of hyperphosphorylation, involving the several isoforms of τ protein, are observed with clinically and pathologically unique manifestations. Most interesting, these other neurodegenerative disorders typically do not have plaques, which are always observed in DAT.21
Plaques, Tangles, and Dementia
Although the combination of Aβ plaques and NFTs was soon recognized as a histopathologic hallmark of DAT, the pathogenesis of the development of the clinical syndrome remained unclear for many years. Even Alzheimer acknowledged that these abnormal structures likely were associated with neuronal dysfunction; however, it was uncertain at the time whether the histopathologic findings were cause or effect.7,8,11,17 To this day, the relationship between plaques and tangles is also incompletely understood. In Alzheimer's second patient, no NFTs were noted, leading some investigators to consider that Aβ pathology may precede and even promote τ pathologies.23,48 To date, however, no proved mechanisms directly link the formation of NFTs to Aβ pathology.37,49
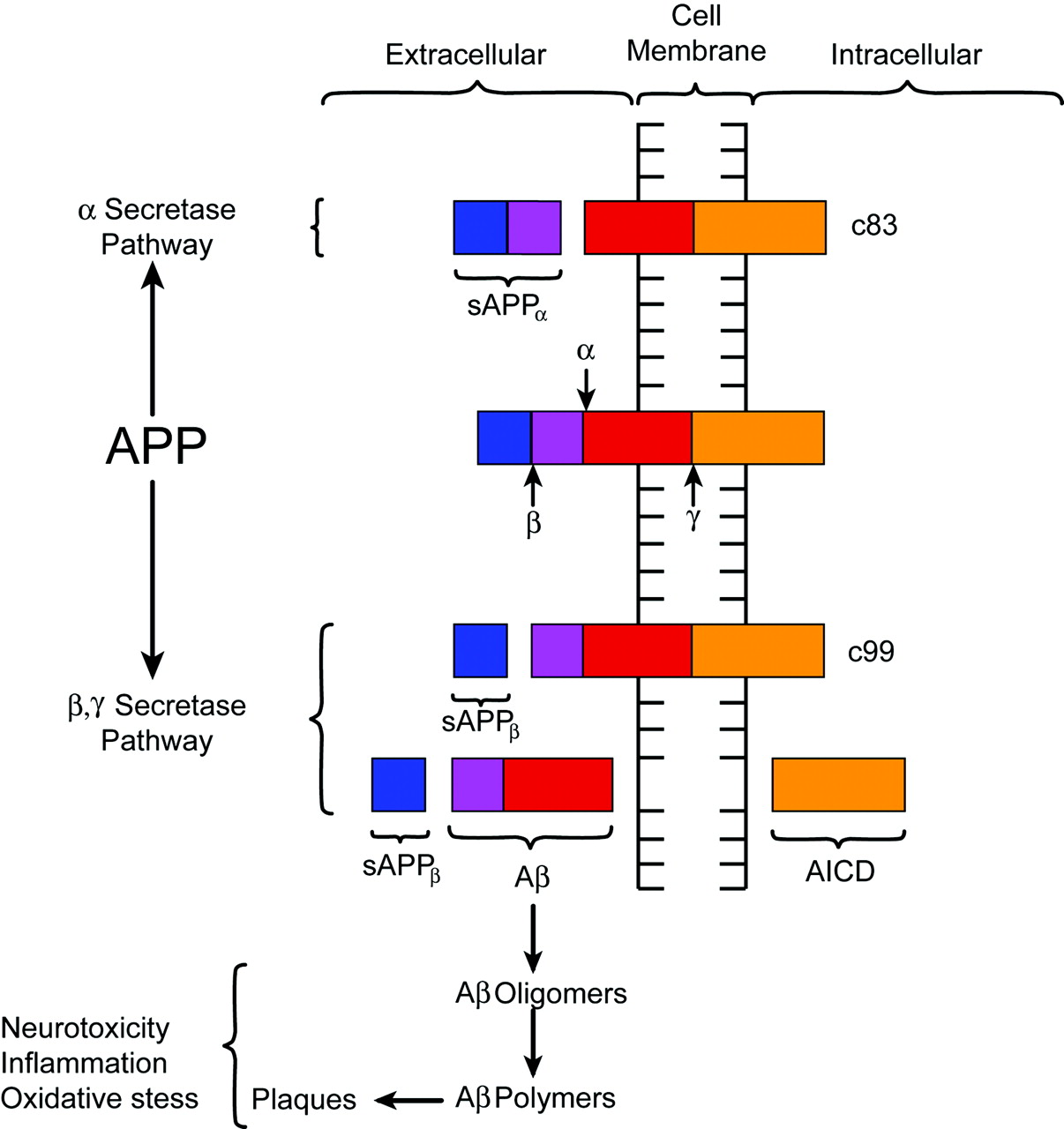
Despite lingering uncertainties, most investigators would agree that the Aβ peptide plays a significant role in the pathogenesis of AD. The Aβ peptide is derived from the metabolism of amyloid precursor protein (APP), a transmembrane glycoprotein processed through endoplasmic reticulum and endosomal and lysosomal pathways and possesses extracellular and intracellular components (Fig 2).23 The prevalence of at least 10 identified isoforms of APP throughout the body suggests a potential role in normal cellular physiology.23,34,45,50 Metabolism of APP can be accomplished through an α-secretase pathway or an alternate and competing β-γ-secretase pathway. The α-secretase pathway entails the cleaving of APP by a protease, known as α-secretase, into an extracellular sAPPα fragment and residual C83 fragment but does not produce Aβ. The sAPPα fragment may actually possess some neuroprotective function. The alternate β-γ-secretase pathway is believed to be problematic in that it generates the Aβ peptide. A protease known as β-secretase cleaves APP, creating an extracellular sAPPβ fragment and a membrane-spanning C99 fragment. The C99 fragment is in turn cleaved by yet another protease known as γ-secretase to form the Aβ peptide, which is ultimately secreted, and the APP intracellular domain (AICD) fragment. The Aβ peptide under certain circumstances, can form highly neurotoxic aggregates consisting of oligomers, which subsequently polymerize into larger fibrillary collections that become plaques.23,37,45 Aβ peptides in the form of oligomers, fibrils, and other species are not only neurotoxic under certain conditions but also proinflammatory and are thought to be involved in oxidative stress. Some investigators have even presented evidence suggesting that Aβ can induce hyperphosphorylation and aggregation of τ protein, but the detailed relationship of amyloid with τ protein and NFTs is not well understood.51

Diagrammatic representation of APP processing. APP is a transmembrane protein with both extra- and intracellular components. APP is processed by 2 competing pathways. The α-secretase pathway generates sAPPα and C83 protein by cleavage of the α-secretase enzyme (α). In the β-secretase pathway, the enzyme β-secretase (β) cleaves APP into an sAPPβ fragment and a C99 fragment. The C99 fragment is further cleaved by γ-secretase enzyme (γ) into an amyloid β fragment (Aβ) and an AICD fragment. The Aβ fragments polymerize. The oligomers and polymers exhibit neurotoxicity. As polymerization proceeds to more complex forms, senile plaques are developed. The C83 fragment is also further processed. However, the function of its products is not fully understood. (The relative sizes of the protein fragments are not drawn to scale.)
Interest in amyloid metabolism and the potential role of Aβ and APP proteins in DAT was stimulated in part by the discovery of genetic anomalies associated with AD.21 For example, mutations in presenilin 1 have been associated with a rare early-onset autosomal dominant familial form of DAT. Presenilin 1 is part of the γ-secretase complex and regulates the cleavage of APP by γ-secretase. Mutations tend to increase the ratio of the Aβ-42 form compared with Aβ-40 variety of protein.41,45,50 Aβ-42, which normally constitutes only 5%–10% of all Aβ peptides, tends to aggregate more aggressively than the Aβ-40 when it is present in higher concentrations.25 Similarly, mutations in a related protein presenilin 2 also cause a form of autosomal dominant AD.41,45,50 Polymorphism of the gene coding for apolipoprotein E, a protein involved in transport and metabolism of lipids, alters one's risk for DAT.23,52 The genetic evidence that links Aβ and APP is further bolstered by predisposition of individuals with trisomy 21 to develop dementia in midlife. These individuals have large numbers of plaques at early ages, thought due to a triple dose of the gene for APP found on chromosome 21 and overexpression of APP.24 Finally, multiple mutations in the APP gene itself cause autosomal dominant familial forms of AD or cerebral amyloid angiopathy. All of these mutations increase total Aβ and Aβ-42 or increase the fibrillogenic properties of Aβ.53
One hypothesis contends that AD begins with a long preclinical stage in which Aβ deposition plays a critical role. As plaques gradually accumulate over the years, their neurotoxic effects produce increasing synaptic and neuronal injury as marked by the development of NFTs. When sufficient brain damage exceeds cognitive reserve, the dementia symptoms begin to appear.47 Also AD-related proteins may be involved in multiple other neuronal functions that are just now being understood, and future discoveries regarding the pathophysiology of AD may shed light on the relationships of amyloid metabolism, plaques, NFTs, and dementia.54
Plaques, Tangles, and Imaging
As mechanisms for the development of AD become better understood, clinical emphasis will increasingly focus on early diagnosis and evaluation of proposed treatments. Attention has recently concentrated on the condition of mild cognitive impairment and its relationship to overt dementia. Limitations of standard psychometric tools have prompted some investigators to consider imaging as a proxy for determining clinical outcomes. Indeed, the National Institute on Aging and the National Institute of Biomedical Imaging and Bioengineering in partnership with several pharmaceutical firms and national foundations have formed the Alzheimer Disease Neuroimaging Initiative to explore new imaging techniques and optimize methods for image acquisition in longitudinal studies.55 In vivo imaging of cerebral amyloid by using molecular probes and PET, volumetric analysis of select portions of the brain with high-resolution MR imaging, and diffusion tensor imaging are just a few of the exciting applications of neuroimaging that will develop in the years to come.56–58
Conclusions
Aβ plaques and NFTs are the histopathologic hallmarks of AD. Plaques are predominately composed of Aβ peptide with frequently associated dystrophic neurites and inflammation. NFTs are intracellular inclusions comprising hyperphosphorylated τ protein that disrupts normal microtubular function. Although the precise pathogenesis of AD is unknown, the burden of these 2 inclusions tends to increase with advancing dementia. Alzheimer's original contribution to the neurosciences resides in his recognition of these 2 structures as pathologic manifestations of a neurodegenerative disorder, rather than the normal and expected culmination of aging.
Footnotes
Supported by National Institute on Aging grants: P50 AG05681 and P01 AG03991.
The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the United States Department of Defense.
References
- Received June 20, 2007.
- Accepted after revision June 27, 2007.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.